
Translate this page into:
Subacute Sclerosing Panencephalitis (SSPE): Experience from a Tertiary-Care Pediatric Center
Meenal Garg, MD Surya Hospitals Tonk Road, Jaipur 302001, Rajasthan India docmeenal@gmail.com
This article was originally published by Thieme Medical and Scientific Publishers Pvt. Ltd. and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Abstract
Introduction Subacute sclerosing panencephalitis (SSPE) is a devastating neurodegenerative disease occurring as a complication of measles infection that is still prevalent in low-resource countries. Clinical and electrographical variability in SSPE can lead to diagnostic delays.
Methods Children diagnosed with SSPE in a tertiary care pediatric hospital in India in a period of 8 years were included in the study. The diagnosis was established on the basis of Dyken's criteria. The demographic data, clinical presentations, investigations, treatment approaches, and outcomes were reviewed and recorded.
Results Thirty-four patients were included in the analysis. Average age at symptom onset was 7 years, 5 months. Majority of the children were not vaccinated for measles. Most patients (80%) presented with stage 2 of illness. Nearly 25% presented with atypical clinical features. Myoclonus was the most predominant feature seen after diagnosis. Electroencephalography (EEG) was the most useful investigation for suspecting the diagnosis. All patients showed deterioration in neurological status with time and 20% died during follow-up.
Conclusion Atypical presentations of SSPE must be recognized in areas with high incidence to institute timely treatment and establish prognosis. EEG findings were found to be the most important indicator for diagnosis. Measles eradication will pave the way for elimination of this dreaded disease.
Keywords
SSPE
subacute sclerosing panencephalitis
measles
neurodegenerative

Introduction
Subacute sclerosing panencephalitis (SSPE) is an acquired neurodegenerative disorder occurring as a complication of measles infection.1 It is characterized by slow progressive cognitive decline followed by motor dysfunction, seizures (predominantly slow myoclonus), vegetative state, and ultimately death. Worldwide incidence of SSPE is ∼4 to 11 cases per 100,000 measles cases.2 Incidence in India is estimated to be 21 cases/million population.3 Several atypical presentations are being recognized in areas with high incidence.4 Although SSPE has pathognomonic clinical and electroencephalography (EEG) features, unusual clinical presentations can pose a diagnostic challenge.
We present an 8-year review of pediatric SSPE patients diagnosed at a tertiary care children hospital in India.
Methods
Children diagnosed with SSPE during January 2011 to December 2018 were included in the study. The diagnosis was established on the basis of Dyken's criteria.5
Demographic, clinical, and diagnostic data was collected retrospectively from hospital records. Patients were clinically classified at the time of presentation per modified Jabbour staging.6 EEG and magnetic resonance imaging (MRI) findings were reviewed.
Measles antibody titers in cerebrospinal fluid (CSF) and serum were performed by enzyme-linked immunosorbent assay (ELISA). CSF to serum titers equal to or greater than 625:1, or rising titer documented via two different samples at least 1 month apart, was considered diagnostic (in-house assay at Neurovirology Laboratory, National Institute of Mental Health and Neurological Sciences, Bengaluru, India). Brain biopsy was not done.
The treatment details were reviewed from hospital data. Follow-up, current status, and outcome were determined from records and current hospital visits. Outcome was classified per modified Rankin score.7 Descriptive data analysis was performed.
Results
Demographics
Thirty-four patients were included. Average age at symptom onset was 7 years, 5 months and at presentation was 7 years, 9 months. Females had lower age of symptom onset and presentation, but this was not statistically significant. Average time from symptom onset to diagnosis was 4 months (range: 1–12 months). In patients with atypical presentation, the time diagnosis was longer by average of 7 months. Nearly 88% children were unvaccinated (Table 1).
|
Demographics |
|
|---|---|
|
Total number, n |
34 |
|
Male: female, n |
22: 12 (1.8: 1) |
|
Average age at symptom onset, years (range) |
7 y, 6 mo (2–13 y) |
|
Males |
7 y, 9 mo |
|
Females |
6 y, 9 mo |
|
Average age at presentation, years (range) |
7y, 9 mo (2y–13 y, 2 mo) |
|
Males |
8 y, 3 mo |
|
Females |
7y, 3 mo |
|
Measles and vaccination |
|
|
Measles, vaccinated |
2 (6%) |
|
Measles, not vaccinated |
29 (85%) |
|
No measles, vaccinated |
2 (6%) |
|
No measles, no vaccination |
1 (3%) |
Clinical Presentation
Majority of children presented with signs and symptoms considered typical of SSPE. One-fourth had atypical features such as generalized tonic–clonic, absence or hypomotor seizures, vision loss, ataxia, dystonia, and parkinsonism. Twenty-seven (80%) patients were in stage 2 at the time of diagnosis. Six patients were in stage 1. Acute fulminant form of SSPE, defined as death or severe neurological impairment within 6 months of symptom onset, was seen in three patients (Fig. 1A, B).
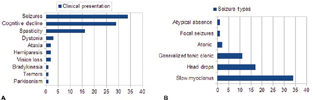
-
Fig. 1 (A) Clinical presentation (Some patients had more than 1 presenting feature). (B) Seizure types (Some patients had more than one seizure types).
Fig. 1 (A) Clinical presentation (Some patients had more than 1 presenting feature). (B) Seizure types (Some patients had more than one seizure types).
One patient with focal cortical dysplasia and epilepsy presented with change in seizure pattern. Two patients had received chemotherapy for Langerhans cell histiocytosis and acute myelogenous leukemia, and had suffered from measles in infancy.
A few patients with confusing presentations are highlighted here.
Case 1
This patient developed subacute-onset dementia and decline in school performance at the age of 11 years. He stabilized and continued to function at subnormal cognitive level for next 3 years but dropped out of school and became emotionally withdrawn. At the age of 14 years, he presented to us with acute onset behavioral abnormalities, psychiatric symptoms, alteration of sleep pattern, enuresis, encopresis, loss of comprehension, slowing of speech, and abnormal gait. There was no history of measles infection or vaccination. He was cooperative but unable to answer simple questions or follow commands. Parkinsonian features were predominant: asymmetric short, shuffling gait with swaying to right side, bradykinesia, cog wheel rigidity, and slow speech. Deep tendon reflexes were brisk with ill sustained ankle clonus. Pronator drift was seen on right side. MRI brain (Fig. 2C, D) showed cerebral atrophy with white matter loss, confluent white matter T2 hyperintense lesions, atrophy of corpus striatum, and mild cerebellar atrophy. EEG showed slow background activity with very occasional bursts of delta range activity. Testing for Wilson disease, adrenoleukodystrophy, Huntington disease, and human immunodeficiency virus was negative. Manganese toxicity was considered in view of exposure to welding fumes but was ruled out. CSF and serum measles antibodies initially tested negative. Child stabilized on antiparkinsonian drugs but did not improve. He further developed asymmetric right-sided chorea. In view of strong suspicion, CSF and serum measles antibody titers were repeated a year later and were found positive.
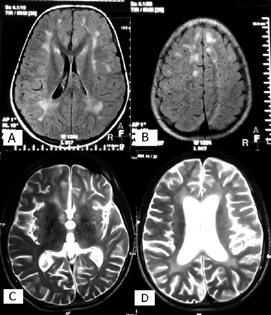
-
Fig. 2 (A, B) T2 fluid-attenuated inversion recovery axial sections of magnetic resonance imaging (MRI) brain showing patchy asymmetrical white matter demyelination in stage 2 of subacute sclerosing panencephalitis (SSPE). (C, D) T2 axial sections of MRI brain showing white matter demyelination, corpus striatal atrophy, and generalized cerebral atrophy in an SSPE patient presenting with parkinsonian features (patient 1 described in the text).
Fig. 2 (A, B) T2 fluid-attenuated inversion recovery axial sections of magnetic resonance imaging (MRI) brain showing patchy asymmetrical white matter demyelination in stage 2 of subacute sclerosing panencephalitis (SSPE). (C, D) T2 axial sections of MRI brain showing white matter demyelination, corpus striatal atrophy, and generalized cerebral atrophy in an SSPE patient presenting with parkinsonian features (patient 1 described in the text).
Case 2
A 7-year-old boy presented with a history of sudden onset imbalance, altered sensorium, visual complaints, and headache. He had history of fever 15 days prior. Papilledema and ataxia were found on examination. MRI brain showed demyelinating changes in cerebral cortex. A diagnosis of acute demyelinating encephalomyelitis (ADEM) was considered. CSF cytology, biochemistry, immunoglobin G index, and oligoclonal bands were normal. EEG was also normal. Child was treated with intravenous pulse corticosteroids without any improvement. His condition rapidly deteriorated during the next 1 month. He developed rigidity and dystonia and became bed bound. He then developed myoclonic jerks of right upper and lower limbs. EEG now showed generalized bursts of delta activity with added spikes and background slowing. CSF measles antibody titers were positive. He was started on antiviral drugs but remained in a vegetative condition and later died. This child presented with acute fulminant form of SSPE and disease course could have been hastened by use of steroids.
Case 3
An 8-year-old girl presented with multiple staring spells (absence seizures). Her examination was normal. EEG (Fig. 3B) showed generalized spike and wave activity at 2.5 to 3 Hz. However, mild slowing of awake background activity was noted on repeat EEGs. MRI brain was normal. Child was started on antiepileptic drugs but seizures persisted. On suspicion of SSPE, CSF and serum measles antibodies were done that were positive. Over next 3 months, she showed mild decline in cognition. Six months later, she developed myoclonic jerks. Subsequent EEGs developed the typical picture of SSPE. Child showed gradual cognitive deterioration.
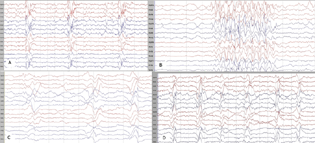
-
Fig. 3 Electroencephalography in four different patients with subacute sclerosing panencephalitis. (A) Pseudoperiodic superimposable generalized epileptiform discharges consisting of spike and wave activity. (B) Generalized spike and wave discharged at a frequency of 2.5 to 3 Hz seen in a patient presenting with absence seizures (patient 3 described in text). (C) Generalized superimposable complexes of delta waves on a very slow background. (D) Generalized pseudoperiodic complexes of delta waves with superimposed sharp waves.
Fig. 3 Electroencephalography in four different patients with subacute sclerosing panencephalitis. (A) Pseudoperiodic superimposable generalized epileptiform discharges consisting of spike and wave activity. (B) Generalized spike and wave discharged at a frequency of 2.5 to 3 Hz seen in a patient presenting with absence seizures (patient 3 described in text). (C) Generalized superimposable complexes of delta waves on a very slow background. (D) Generalized pseudoperiodic complexes of delta waves with superimposed sharp waves.
EEG
The most consistent EEG findings were slowing of background activity (n = 29, 85%) and superimposable delta bursts (n = 25, 73%). Periodic to pseudoperiodic epileptiform activity clinically corresponding to myoclonic jerks was seen in 50%. Focal epileptiform activity was seen in half, most common being frontal discharges. Other EEG findings were multifocal epileptiform discharges (n = 5, 15%), generalized spike, and slow wave discharges (n = 7, 20%) and generalized sharps and slow waves (n = 11,32%). Nearly all EEGs evolved to a typical pattern of SSPE within 3 months of presentation (Fig. 3).
MRI Brain
MRI brain was performed in 27 (79%) patients. Diffuse or focal T2/fluid-attenuated inversion recovery white matter hyperintensities mainly in temporo-parieto-occipital region was the most common finding. In late stage, generalized cerebral atrophy and periventricular white matter changes were seen (Table 2; Fig. 2).
|
MRI findings |
n (%) |
|---|---|
|
MRI done in total patients |
27/34 (79%) |
|
➢ Normal |
13/27 (48%) |
|
➢ White matter changes (diffuse/focal) |
12/27 (44%) |
|
Temporo-parieto-occipital |
3 (15%) |
|
Patchy, asymmetrical |
8 (40%) |
|
➢ Thalamic/cerebellar involvement |
2 (10%) |
|
➢ Cortical atrophy |
2(7.4%) |
|
➢ Focal encephalitis |
1 (4%) |
|
➢ Multiple infarcts |
1(4%) |
Abbreviation: MRI, magnetic resonance imaging.
Others
Testing for human immunodeficiency virus (ELISA) was negative in all patients.
Treatment
All patients were started on antiviral drugs (Fig. 4), out of which only isoprinosine was continued in all patients. One patient received intrathecal interferon therapy. Patients were on one to five antiepileptic drugs (average: 2). High-dose levetiracetam was the most commonly used drug for myoclonus (n = 26, 76.5%). Other drugs were valproate (n = 24), clobazam (n = 12), zonisamide (n = 3), carbamazepine (n = 2), clonazepam (n = 4), topiramate (n = 2), and lamotrigine (n = 1). Patients reported subjective benefit with levetiracetam and sodium valproate in seizure reduction.
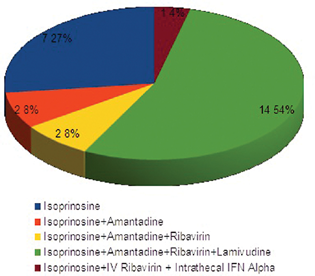
-
Fig. 4 Graphical presentation of treatment: antiviral drugs.
Fig. 4 Graphical presentation of treatment: antiviral drugs.
Outcome
The mean duration of follow-up was 15 months (range: 1–60 months). All patients showed deterioration in their neurological status over time with 6 (18%) in stage 4 of modified Rankin scale and 21 (62%) in stage 5. Seven patients (20%) had died at the end of follow-up.
Discussion
Measles is presently targeted for elimination in India. The relatively large number of patients seen at single pediatric care center in this study may indicate a high burden of SSPE in our country. The patients included in this study largely belonged to the state of Maharashtra, but a sizable number traveled from distant places with or without the final diagnosis of SSPE. Thus, under-diagnosis and under-reporting of SSPE are likely in our setting due to lack of awareness and limited diagnostic services.
A male preponderance was noted in our study group, which has been reported previously.8 The lower age of affection in females could be because of poor health services and immunization facilities available to female children in many parts of India. Most of the patients in our study were not immunized and had suffered from measles. The percentage of unvaccinated children was higher as compared with other Indian studies.9 This can be due to preponderance of rural patients and poor reach of vaccination programs.
The clinical presentation of our patients largely corresponded to the classical features seen in SSPE.10 However, nearly 25% patients were seen with atypical features. Erturk et al11 in their study have reported that 17% of patients with SSPE had atypical presentations. Atypical features may include isolated psychiatric manifestations, poorly controlled seizures, isolated extrapyramidal symptoms, and stroke-like illness. Clinical presentations resembling ADEM,12 with parkinsonian features13 and absence seizures,14 as in our patients, have been reported rarely. Based on our data, we can suggest that a relatively low threshold for suspecting SSPE should be practiced in areas with high incidence of measles infection. The average diagnostic delay in our study was found to be around 4 months and was higher for patients with uncommon symptoms. Subtle symptoms in early stages, atypical clinical features, and delayed referral to specialists can contribute to diagnostic delay.
The characteristic EEG in SSPE consists of stereotyped, bilaterally synchronous, and symmetrical superimposable periodic complexes sometimes with intermingled spikes or sharp waves.15 Although typical EEG features are a part of Dyken's criteria, mild abnormalities on EEG were quite helpful for us in suspecting the disease. A knowledge of atypical EEG characteristics may raise early suspicion and shorten the time to diagnosis.16 Neuroimaging is primarily helpful in excluding other differentials as there is no characteristic picture in SSPE.
No standard treatment guidelines are available. The mainstay of treatment is the use of antiepileptic drugs for seizure control and antiviral agents with or without immunomodulators. We found that while drugs prolong longevity, they did not offer substantial clinical improvement. The financial constraints and risk of infection outweighed the possible benefits of intraventricular interferon-α for our patients. Inosine pranobex has been found to be the most beneficial therapy in patients with SSPE in various studies.17 18 Based on limited evidence, several other antiviral drugs were also tried. There was no clear superiority of any treatment and all patients eventually showed neurological deterioration. Generalized seizures responded well to antiepileptic drugs. High-dose levetiracetam resulted in subjective improvement in myoclonus initially. A few reports have also highlighted the efficacy of levetiracetam in SSPE.19 20 However, myoclonus was relentlessly progressive.
Our study had many limitations. The data are based on our single-center experience. Being a tertiary center, there is likely a referral bias and exact incidence of the disease cannot be determined. Our population was a heterogeneous one, which can be expected for rare neurodegenerative disorders. There was variation in treatment offered depending on the clinical condition, financial status of patient, and preference of the treating physician. Much of the data was collected retrospectively from the records.
Conclusion
SSPE remains a life-threatening complication of measles. This single-center study highlights disease burden and variability of clinical presentation. A high index of suspicion can lead to timely diagnosis and proper counseling. Effective vaccination strategy is the only measure to decrease the disease burden in the community.
Conflict of Interest
None declared.
Funding None.
References
- Subacute sclerosing panencephalitis: more cases of this fatal disease are prevented by measles immunization than was previously recognized. J Infect Dis. 2005;192(10):1686-1693.
- [Google Scholar]
- Review of the effect of measles vaccination on the epidemiology of SSPE. Int J Epidemiol. 2007;36(6):1334-1348.
- [Google Scholar]
- Atypical presentations of SSPE: a clinical study in four cases. Turk J Pediatr. 2007;49(3):295-300.
- [Google Scholar]
- Subacute sclerosing panencephalitis. Current status. Neurol Clin. 1985;3(1):179-196.
- [Google Scholar]
- Subacute sclerosing panencephalitis presenting as simple partial seizures. J Child Neurol. 1991;6(2):146-149.
- [Google Scholar]
- Interobserver agreement for the assessment of handicap in stroke patients. Stroke. 1989;20(6):828. (letter)
- [Google Scholar]
- Subacute sclerosing panencephalitis: a clinical appraisal. Ann Indian Acad Neurol. 2013;16(4):631-633.
- [Google Scholar]
- Subacute sclerosing panencephalitis: an update. Dev Med Child Neurol. 2010;52(10):901-907.
- [Google Scholar]
- Subacute sclerosing panencephalitis (SSPE) presenting as acute disseminated encephalomyelitis in a child. J Child Neurol. 2009;24(7):899-903.
- [Google Scholar]
- Parkinsonian presentation of SSPE: report of two cases. Neurol Asia. 2008;13:117-120.
- [Google Scholar]
- Subacute sclerosing panencephalitis (SSPE): an insight into the diagnostic errors from a tertiary care university hospital. J Child Neurol. 2007;22(6):683-688.
- [Google Scholar]
- Atlas of Pediatric Electroencephalography. (2nd edition.). Philadelphia, PA: Lippincott-Raven; 1999. 370.24
- [Google Scholar]
- Atypical EEG findings in subacute sclerosing panencephalitis. Clin Neurophysiol. 2005;116(8):1762-1767.
- [Google Scholar]
- Long-term follow-up of patients with subacute sclerosing panencephalitis treated with inosiplex. Ann Neurol. 1982;11(4):359-364.
- [Google Scholar]
- Randomized treatment study of inosiplex versus combined inosiplex and intraventricular interferon-α in subacute sclerosing panencephalitis (SSPE): international multicenter study. J Child Neurol. 2003;18(12):819-827.
- [Google Scholar]
- Successful treatment of encephalopathy and myoclonus with levetiracetam in a case of subacute sclerosing panencephalitis. J Child Neurol. 2009;24(6):763-767.
- [Google Scholar]
- Suppression of post-hypoxic and post-encephalitic myoclonus with levetiracetam. Neurology. 2001;56(3):411-412.
- [Google Scholar]




