
Translate this page into:
Phenotypic features of epilepsy due to sodium channelopathies – A single center experience from India
-
Received: ,
Accepted: ,
How to cite this article: Viswanathan LG, Alapati S, Nagappa M, Mundlamuri R, Kenchaiah R, Asranna A, et al. Phenotypic features of epilepsy due to sodium channelopathies – A single center experience from India. J Neurosci Rural Pract 2023;14:603-9.
Abstract
Objectives:
Nearly 40% of pediatric epilepsies have a genetic basis. There is significant phenotypic and genotypic heterogeneity, especially in epilepsy syndromes caused by sodium channelopathies. Sodium channel subunit 1A (SCN1A)-related epilepsy represents the archetypical channel-associated gene that has been linked to a wide spectrum of epilepsies of varying severity. Subsequently, other sodium channels have also been implicated in epilepsy and other neurodevelopmental disorders. This study aims to describe the phenotypes in children with sodium channelopathies from a center in Southern India.
Materials and Methods:
This is a retrospective, descriptive, and single-center study. Out of 112 children presenting with epilepsy who underwent genetic testing between 2017 and 2021, 23 probands (M: F = 12:11) were identified to have clinically significant sodium channel mutations. Clinical presentation, electroencephalography, and imaging features of these patients were recorded. The utility of genetic test results (e.g., in planning another child, withdrawal of medications, or change in treatment) was also recorded.
Results:
Age at onset of seizures ranged from day 4 of life to 3.5 years. Clinical epilepsy syndromes included generalized epilepsy with febrile seizures plus (n = 3), Dravet syndrome (n = 5), early infantile epileptic encephalopathy (n = 7), drug-resistant epilepsy (n = 5), and epilepsy with associated movement disorders (n = 3). The most common type of seizure was focal with impaired awareness (n = 18, 78.2%), followed by myoclonic jerks (n = 8, 34.78%), epileptic spasms (n = 7, 30.4%), bilateral tonic-clonic seizures/generalized tonic–clonic seizures (n = 3, 13%), and atonic seizures (n = 5, 23.8%). In addition to epilepsy, other phenotypic features that were discerned were microcephaly (n = 1), cerebellar ataxia (n = 2), and chorea and dystonia (n = 1).
Conclusion:
Sodium channelopathies may present with seizure phenotypes that vary in severity. In addition to epilepsy, patients may also have other clinical features such as movement disorders. Early clinical diagnosis may aid in tailoring treatment for the given patient.
Keywords
Pediatric epilepsy
Channelopathy
Sodium channel epilepsy
Dravet syndrome
Generalized epilepsy with febrile seizures plus
Epilepsy genetics

INTRODUCTION
Voltage-gated sodium channels have a vital role in the functioning of neurons. The channel essentially consists of a pore-forming alpha subunit and subsidiary beta-subunit(s).[1] The ion conductance of these channels dictates neuronal functions in many types of neurons. Sodium channel subtypes have unique distributions in the central (Nav1.1, Nav1.2, and Nav1.3 subtypes) and peripheral nervous systems (Nav1.6). Clinical manifestations vary according to the subunit types that harbor mutations.[2] SCN1A-related epilepsy syndromes span a spectrum of phenotypes ranging from benign age-limited epilepsies to devastating neurodevelopmental syndromes such as Dravet syndrome (DS). Various other sodium channel subunits have also been characterized to have a similar spectrum such as SCN1A although a touch more uncommon. With the advent of whole exome sequencing, knowledge of these rarer phenotypes has evolved significantly. We aimed to study the phenotypic expressions of sodium channel mutations that were identified at our center in this study.
MATERIALS AND METHODS
This was a retrospective study conducted at a tertiary care center in Southern India. The study was approved by the Institutional Ethics Committee.
One hundred and twelve children presenting with epilepsy had undergone genetic testing after informed consent from 2016 to 2021. Genetic testing was done when it was felt to be clinically appropriate by the treating epileptologist. Twenty-three patients were identified to have pathogenic/significant variants in sodium channel subunits. Clinical findings and treatment details on the number of anti-seizure medications, use of vitamins/steroids, and or other treatment modalities (such as ketogenic diet/surgery) were also documented. Imaging studies were reviewed in all included subjects.
Electrophysiological data in the form of electroencephalography (EEGs) were reviewed for background (normal/slow/asymmetric) and sleep transients. Interictal discharges were qualified based on standard International Federation for Clinical Neurophysiology (IFCN) definitions. Pertinent details from exome sequencing reports such as type of variation (missense, non-sense, deletion, duplication, frameshift, and insertions), zygosity, in silico predictions, and their classification according to the American College of Medical Genetics were gathered.
RESULTS
Clinical and genotype
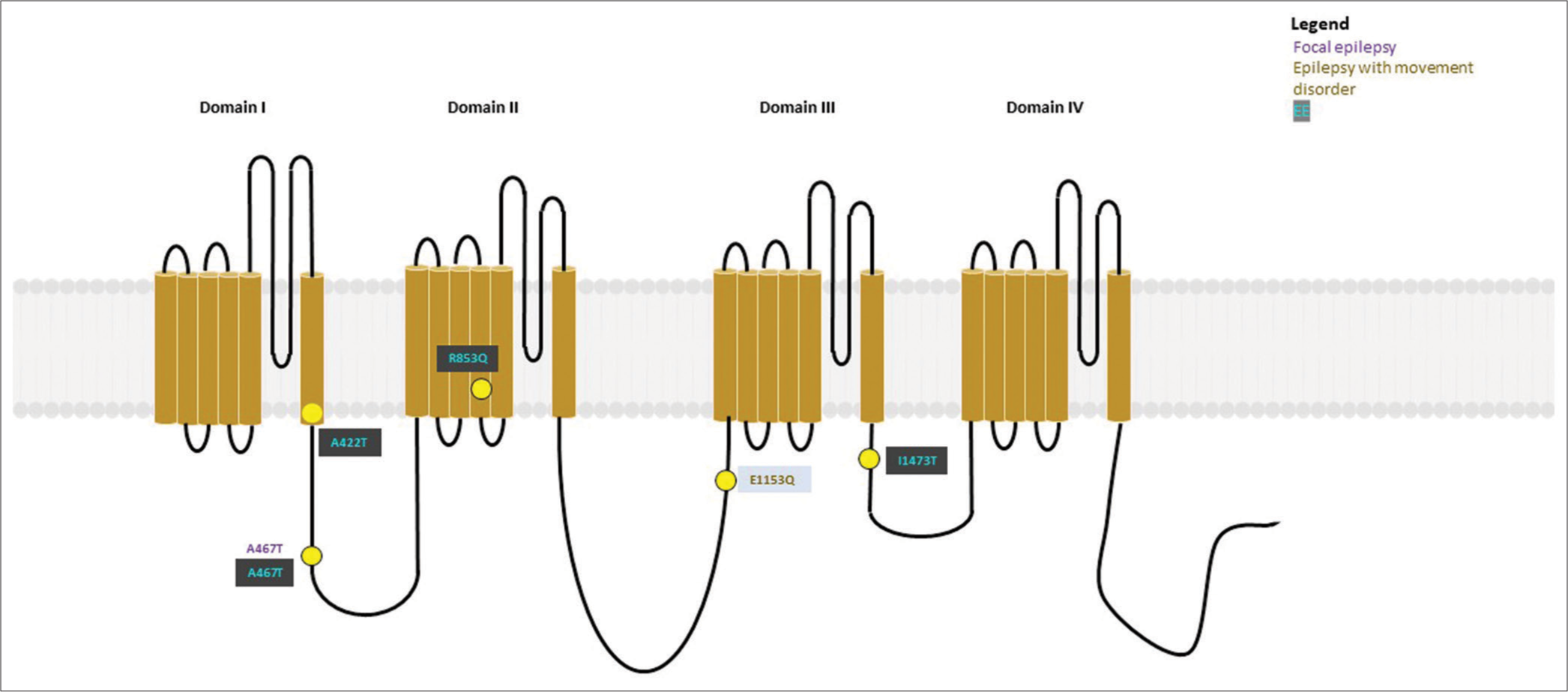
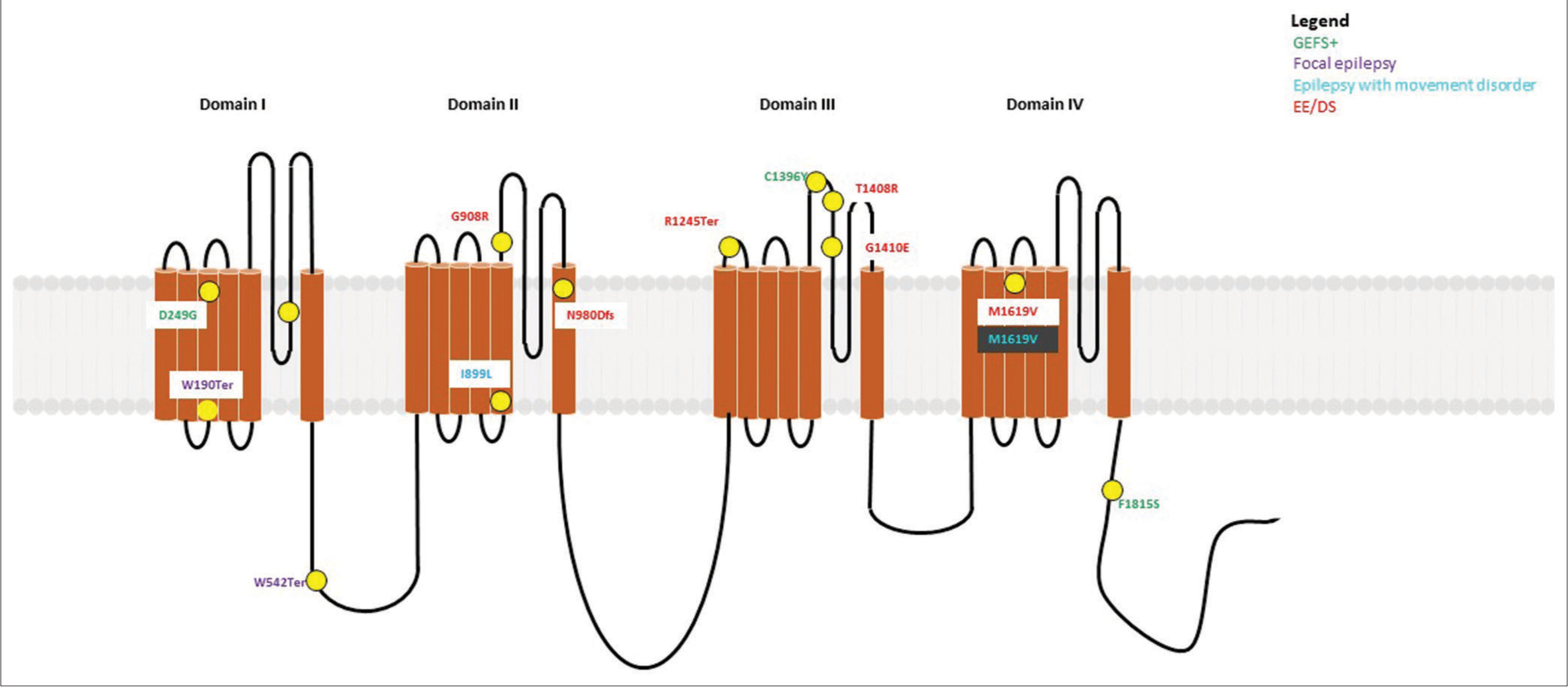
Both sexes were equally affected (M: F = 12:11). The mean age at onset was 10.4 ± 12 months (range: 0.03–4 years, median 6 months). Fever/vaccine-induced seizures were observed in 10 probands (43.4%). Family history of epilepsy in first-degree relatives was noted in two probands. Focal seizures with impaired awareness (n = 18, 78.2%) were the most common phenotype, followed by myoclonic jerks (n = 8, 34.78%) and infantile spasms (n = 7, 30.4%). In addition, focal to bilateral tonic–clonic seizures/generalized tonic–clonic seizures (n = 3, 13%) and atonic seizures (n = 5, 23.8%) were also observed. Multiple semiologies were present in 73.9% of the cohort (n = 17). Epileptic encephalopathies (EEs) such as early infantile forms (n = 5, 38%), West syndrome (n = 2, 9.5%), Lennox-Gastaut syndrome (n = 1, 4.7%), and DS constituted the most common presentations (n = 5, 38%) of SCN1A variants. Other phenotypic features observed were microcephaly (n = 1), cerebellar ataxia in two subjects, and chorea-dystonia in one proband. Developmental abnormalities were present in 18 children. Three probands with generalized epilepsy with febrile seizures plus (GEFS+), and P15 and 22 with refractory focal epilepsy had normal development. In our study, all variants were found in a heterozygous state that the most common subunit with mutations was SCN1A (n = 13, 56.52%), followed by SCN2A (n = 6, 28.5%). Mutation in SCN8A was detected in two probands (8.6%) and SCN1B in one (4.7%). Among the probands with SCN1A mutations, eight (61%) carried reported pathogenic variants and the remaining were novel. Four probands were identified to have non-sense variants, among which one was a novel variant. Most of the mutations were missense (n = 9). Details of mutations and patient characteristics are shown in Table 1, Figures 1 and 2. Five probands with SCN1A had DS and four had drug-resistant focal seizures. Three children had a GEFS+-like phenotype and one had early infantile EE. Two patients (one with DS and the other with EIEE) shared the same mutation in SCN1A. Phenotypic heterogeneity was also noted in SCN2A. Two probands carrying the same variant presented with different phenotypes. One presented with a drug-resistant focal epilepsy while the other developed early infantile EE akin to Ohtahara syndrome. All patients carrying SCN2A variants had severe phenotypes and adverse neurodevelopmental profiles.
| Patient | Age at onset | Sex | Development | Fever Triggered Seizures | Clinical Phenotype | Gene | Location (exon) | DNA | Aminoacid |
|---|---|---|---|---|---|---|---|---|---|
| P1 | 3 | F | DR | + | DS | SCN1A | 19 | c.3733C>T | p.Arg1245Ter |
| P2 | 6 | M | DD | + | DS | SCN1A | 21 | c.4222T>C | p.Trp1408Arg |
| P3 | 12 | F | R | + | DS | SCN1A | 26 | c.4855A>G | p.Met1619Val |
| P4 | 9 | M | N | + | GEFS+ | SCN1A | 28 | c.5444T>C | p.Phe1815Ser |
| P5 | 3 | F | N | + | GEFS+ | SCN1A | 21 | c.4187G>A | p.Cys1396Tyr |
| P6 | 4 | M | DD | + | DS | SCN1A | 15 | c.2930_2936 Dup | p.Asn980 AspfsTer19 |
| P7 | 6 | M | DD | + | DS | SCN1A | 21 | c.4229G>A | p.Gly1410Glu |
| P8 | 48 | F | N | - | GEFS+ | SCN1A | 9 | c.746A>G | p.Asp249Gly |
| P9 | 3 | M | DR | - | EIEE | SCN2A | 10 | c.1264G>A | p.Ala422Thr |
| P10 | 8 | M | DR | - | WS | SCN2A | 15 | c.2558G>A | p.Arg853Gln |
| P11 | 5 | F | DR | - | WS | SCN1A | 26 | c.4855A>G | p.Met1619Val |
| P12 | 0.3 | M | DD | - | EIEE | SCN2A | 24 | c.4418T>C | p.Ile1473Thr |
| P13 | 1.5 | F | DR | - | Epilepsy with chorea and dystonia | SCN1A | 18 | c.2695A>C | p.Ile899Leu |
| P14 | 4 | M | DR | + | Epilepsy with ADHD | SCN1A | 10 | c.1624C>T | p.Arg542Ter |
| P15 | 40 | M | N | - | Focal epilepsy | SCN1B | 3 | c.595C>T | p.Pro199Ser |
| P16 | 21 | M | DD | - | EE | SCN2A | 11 | c.1399G>A | p.Ala467Thr |
| P17 | 18 | F | DD | + | Epilepsy with ataxia | SCN2A | 18 | c.3457G>C | p.Glu1153Gln |
| P18 | 30 | M | R | - | Refractory epilepsy | SCN2A | 11 | C.1399G>A | p.Ala467Thr |
| P19 | 6 | F | DR | - | EIEE | SCN8A | 27 | c.5615G>A | p.Arg1872Gln |
| P20 | 1 | M | DD | - | Refractory Epilepsy | SCN1A | 8 | c.1166A>C | p.Gln389Pro |
| P21 | 6 | F | DD | - | EIEE | SCN1A | 15 | c.2722G>C | p.Gly908Arg |
| P22 | 3 | F | N | + | Refractory epilepsy | SCN1A(-) | 4 | c.569G>A | p.Trp190Ter |
| P23 | 3 | F | DR | - | Epilepsy with IDD | SCN8A(+) | 25 | c.4423G>A | p.Gly1475Arg |
DD-developmental delay, DR-delay with regression, R- regression only, N normal, DS Dravet syndrome, EIEE early infantile epileptic encephalopathy, IDD intellectual and developmental disabilities
- Variants (n = 13) identified in SCN1A have been depicted as yellow dots. Colored text in the boxes represents various phenotypes as described in the figure.
- Variants (n = 5) identified in SCN2A have been depicted as yellow dots. Colored text in the boxes represents various phenotypes as described in the figure.
EEG and imaging features
Background slowing was evident in all patients with DS and EE. Epileptiform discharges were multifocal in 4 subjects (17.4%) and generalized discharges were identified in up to 57% (n = 12). Classical hypsarrhythmia pattern was not identified in any of the subjects with West Syndrome/Early infantile epileptic encephalopathy (WS/EIEE).
Magnetic resonance imaging (MRI) was available for all patients and was normal in 60% of subjects (n = 14). Non-specific periventricular white matter changes (n = 6, 28.5%), cerebral (n = 1)/cerebellar (n = 1), and atrophy (n = 6, 28.5%) were the commonly detected abnormalities. Central tegmental tract hyperintensity was noted in one proband. No malformations of cortical development were detected. A clinical example is shown in Figure 3.
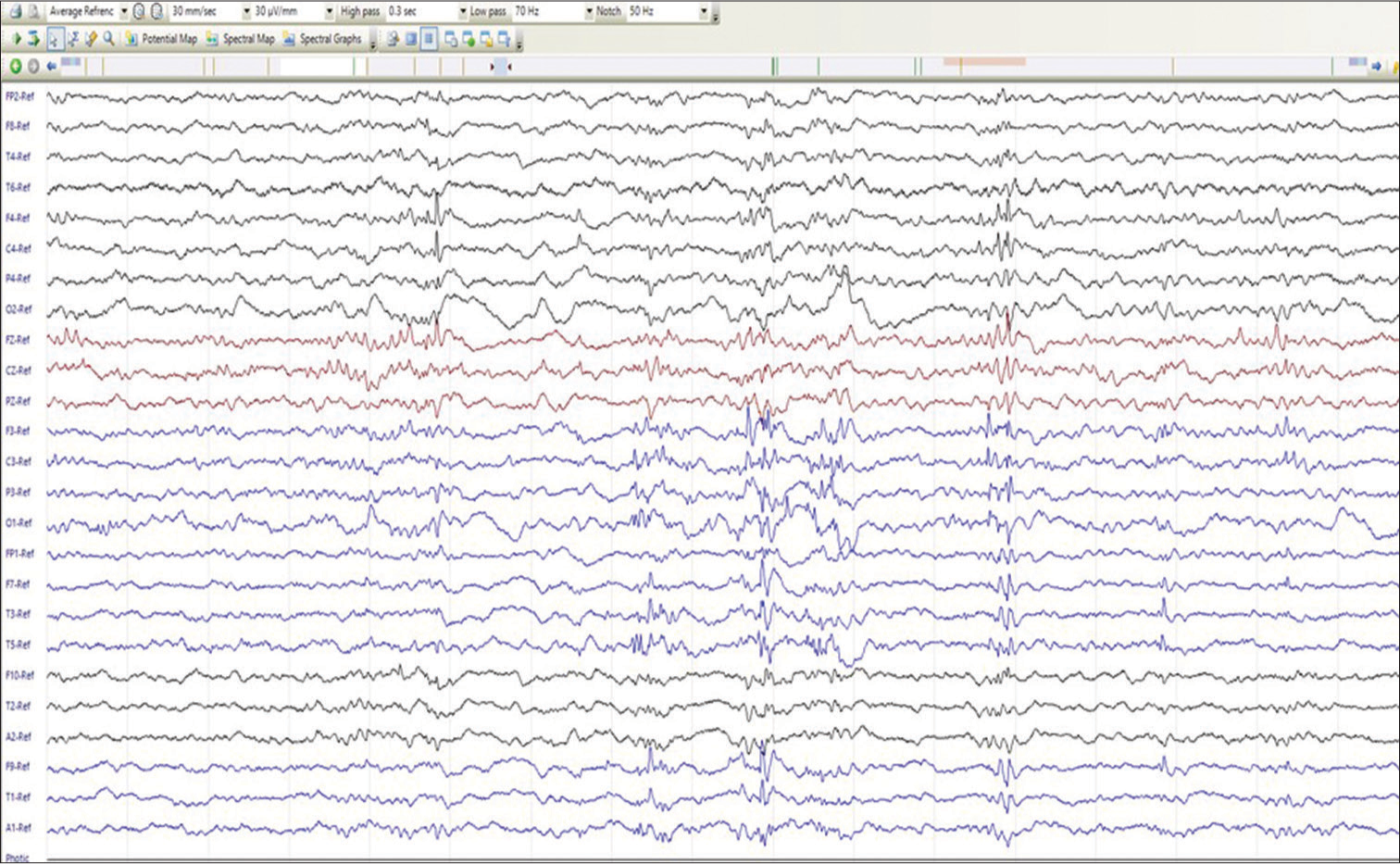
- Sleep electroencephalography of P2 (Dravet syndrome) showing polyspike–spike wave discharges from the left hemispheric leads. Furthermore, note a solitary spike-wave discharge from the right frontocentral region (F4 and C4).
Treatment and clinical implications
Four patients were administered sodium channel blockers for variable durations (carbamazepine = 3 and lamotrigine = 1) before genetic testing. Among them, one proband with DS harboring a frameshift mutation in SCN1A had a drastic increase in the frequency of seizures necessitating the stoppage of treatment. The other two probands did not have worthwhile seizure reduction. The most common ASMs used in this cohort were levetiracetam and valproate. Two patients with DS were also administered stiripentol which resulted in a slight improvement in seizure frequency. Worsening of seizure frequency (tonic spasms) was noted with vigabatrin in one proband with SCN1A mutation R853Q.
Four out of 21 patients were administered steroids (IV methylprednisolone or adrenocorticotropic hormone). These patients had early infantile EE. Response to steroids was noted but short-lasting and no child had complete remission from seizures. One proband presented with refractory focal seizures with multiple episodes of status epilepticus. Ictal EEG was suggestive of right posterior quadrant onset and MR imaging was normal. Further pre-surgical workup was abandoned as a non-sense mutation (p.Trp190Ter) was identified on exome sequence.
DISCUSSION
Channelopathies constitute an important paradigm for genetic epilepsies. In this study, we identified mutations in SCN1A, 1B, 2A, and 8A across a wide spectrum of epilepsy phenotypes affecting children right from early infancy to 3.5 years of age. Fever-triggered seizures which are quite common in sodium channelopathies were noted in 75% of patients, which parallels a finding that was also reported in another study. Five children presented with phenotypic features of DS and all had mutations in SCN1A. Loss of function mutations in SCN1A are seen in up to 80% of patients with DS. Deleterious missense (n = 3) and non-sense (n = 2) were detected in the affected subjects of this study. Ceuleman et al. hypothesized that truncated proteins due to frameshift and non-sense variants would invariably result in a severe phenotype as it would adversely affect channel function.[3] It is hypothesized that the truncated protein exerts a dominant negative effect on the wild-type allele resulting in abnormal sodium channel function.[4,5] Missense mutations, on the other hand, may lead to haploid insufficiency resulting in disease. Brunklaus et al. commented that the type of SCN1A mutation may not have a critical bearing of the phenotype or severity of DS.[6] In this study, non-sense mutations were also observed in other phenotypes. P22 (c.569G>A[p.Trp190Ter]) suffered from drug-resistant focal epilepsy. She was evaluated as a possible candidate for epilepsy surgery. Plans for invasive EEG were deferred after the genetic diagnosis was made. A series published by Skjei et al. characterized the outcomes in children with SCN1A mutations who underwent epilepsy surgery.[7] Half of those probands were noted to have normal findings on MRI and the rest had mesial temporal sclerosis. Surgical resection did not improve the frequency of seizures. Intuitively, one may be concluded that diffuse cellular physiological changes induced by SCN1A mutants would obviate focal brain resection. Comparable results were noted by Venezyroglou et al.[8] Apart from epilepsy, SCN1A has also been implicated in certain movement disorders. Proband (P13) carrying c.2695A>C (p.Ile899Leu) suffered from epilepsy, chorea and dystonia, developmental delay, and regression. Sadleir et al. have described a similar phenotype due to a mutation in exon 5 (c.677C>T, p.Thr226Met).[9] Hyperkinetic movement disorders are not rare in SCN1A mutations. Due to the coexistence with complex seizure semiologies, these may be difficult to distinguish from epilepsy in a few cases.
Developmental encephalopathy with profound intellectual impairment can also be seen in SCN1A-related disorders. P11 presented with infantile spasms and went on to have multiple seizure semiologies, similar to the results described by Gowda. P14 (c.1624C>T[p.Arg542Ter]) presented with autism and refractory epilepsy and regression of milestones.[10] Till et al. had reported a proband with DS with the same variant.[11] Grantham scores of variants that cause GEFS+ tend to be lower than those causing developmental and epileptic encephalopathy (DEE) and DS. More severe phenotypes thus occur due to amino acid substitutions that harm proper ion channel function.
The two missense novel variants in this study include c.4222T>C (p.Trp1408Arg) variant and c.4229G>A (p.G1410E), respectively. They were found to be in silico damaging by SIFT, polyphen 2, and mutation taster. Both these mutations were in domain 3 of the channel in close relation to the inactivation gate between domain III and IV and could explain its damaging effect.
MRI was abnormal in only 3 patients (23%). Changes including atrophy and white matter changes were observed. Similarly, Guerrini et al. also described normal imaging in most patients only a few showed abnormalities such as hippocampal sclerosis, focal brain atrophy, and cortical dysplasia.[12] These abnormalities were however not detected in our cohort.
SCN2A mutations also present with a wide clinical spectrum including GEFS plus, DS, acute encephalopathy with refractory, repetitive partial seizures, autism, and severe IDD.[13-17] In this study, six patients carried SCN2A mutations; all of which were missense. Two probands P10 presented with EE and harbored reported pathogenic variant.[16] P16 and 18 carried that same missense novel variant C.1399G>A (p.Ala467Thr). Both probands had presented with a similar phenotype characterized by refractory epilepsy with developmental delay. This variant is located on the connecting link between the alpha subunits (domains 1 and 2), functional implication of this variant causes protein instability and has been described with phenotype having an intellectual disability without epilepsy in the study by Howell et al.[18] and as DEE in the study by Reynolds et al.[19] these were different as compared to phenotype in this study. P17 (c.3457G>C [p.Glu1153Gln]) presented with epilepsy associated with ataxia. A similar phenotype has been described in subjects carrying variants: c.5644C>G(p. R1882G), c.4565G>C (p.G1522A), c.788C>T(p.A263V), and c.788C>T(p.A263V) as shown in Schwarz et al.[20] These patients usually have disease onset between 15 months and 3.7 years. It was hypothesized that cerebellar ataxia in part could be due to the upregulation of Na1.2 in cerebellar granule cells.
Imaging of these patients revealed only non-specific findings such as mild atrophy or subtle white matter hyperintensities in periventricular areas in most patients. Other abnormalities such as cerebral or cerebellar atrophy, thin corpus callosum,[15] dentate-olivary dysplasia,[21] posterior perisylvian, and bilateral parietal polymicrogyria[22] have been described in SCN2A-related disorders. The role of the sodium channels in the migration and maturation of oligodendroglial progenitor cells may explain the occurrence of such abnormalities.[23]
Mutations in SCN8Aand SCN9A may result in epilepsy and other neurodevelopmental disorders including autism. More than 150 additional de novo missense mutations have now been reported to date.[24,25]
In this study known (P19) and one novel (P23), missense variants have been recorded.[26]
P19: (c.5615G>A[p.Arg1872Gln]) presented with EE. Larsen et al. described the same variant with a DS-like phenotype, moderate IDD, seizures of multiple semiologies, autistic features, and motor stereotypies.[27] P23 (c.4423G>A[p. Gly1475Arg]) presented with epilepsy and IDD. The proband also had clusters of seizures followed by regression of milestones. It has been noted that SCN8A mutants almost always cause moderate-to-severe IDD and/or autistic traits. The reported variant at position 1872 is an emerging hotspot. Most SCN8A are gain of function mutations and sodium channel blockers may be a useful therapeutic option to control seizures in these individuals.
Although SCN1B was the first gene associated with GEFS+ to date, only a few variants are reported in the literature.[28] In this study, P15 carried c.595C>T(p.Pro199Ser) missense variant presented with epilepsy (focal seizures), with normal development. Interestingly, this patient also had central tegmental tract hyperintensities which were restricting. A GEFS+-like phenotype and myoclonic epilepsy have been associated with SCN1B mutations. Fenfluramine has been found to be of benefit in cases with difficult-to-control seizures.[29]
CONCLUSION
This study highlights a few key aspects of the genotype – phenotype spectrum of sodium channelopathies. Early recognition of epilepsy phenotypes may aid in choosing/avoiding certain anti-seizure medications. A few of these patients may also present with refractory focal epilepsy. Appropriately used genetic testing methods may avoid pre-surgical workup/epilepsy surgery in these patients as postoperative outcomes in such epilepsies are usually poor. An early genetic diagnosis may aid in equipping both the treating clinician and the family with more information for better management of epilepsy and associated conditions.
Ethical approval
The authors declare that they have taken the ethical approval from IRB/IEC.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship
Nil.
References
- From ionic currents to molecular mechanisms: The structure and function of voltage-gated sodium channels. Neuron. 2000;26:13-25.
- [CrossRef] [PubMed] [Google Scholar]
- International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol Rev. 2005;57:397-409.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical correlations of mutations in the SCN1A gene: From febrile seizures to severe myoclonic epilepsy in infancy. Pediatr Neurol. 2004;30:236-43.
- [CrossRef] [PubMed] [Google Scholar]
- Generalized epilepsy with febrile seizures plus (GEFS+) spectrum: Clinical manifestations and SCN1A mutations in Indonesian patients. Epilepsy Res. 2010;90:132-9.
- [CrossRef] [PubMed] [Google Scholar]
- De novo SCN1A mutations are a major cause of severe myoclonic epilepsy of infancy. Hum Mutat. 2003;21:615-21.
- [CrossRef] [PubMed] [Google Scholar]
- Prognostic, clinical and demographic features in SCN1A mutation-positive Dravet syndrome. Brain. 2012;135:2329-36.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical and histopathological outcomes in patients with SCN1A mutations undergoing surgery for epilepsy. J Neurosurg Pediatr. 2015;16:668-74.
- [CrossRef] [PubMed] [Google Scholar]
- Focal epilepsy in SCN1A-mutation carrying patients: Is there a role for epilepsy surgery? Dev Med Child Neurol. 2020;62:1331-5.
- [CrossRef] [PubMed] [Google Scholar]
- Not all SCN1A epileptic encephalopathies are Dravet syndrome: Early profound Thr226Met phenotype. Neurology. 2017;89:1035-42.
- [CrossRef] [PubMed] [Google Scholar]
- Case series of early SCN1A-related developmental and epileptic encephalopathies. J Pediatr Neurosci. 2021;16:212-7.
- [CrossRef] [PubMed] [Google Scholar]
- Mutation spectrum of the SCN1A gene in a Hungarian population with epilepsy. Seizure. 2020;74:8-13.
- [CrossRef] [PubMed] [Google Scholar]
- Neuroimaging and neuropathology of Dravet syndrome. Epilepsia. 2011;52(Suppl 2):30-4.
- [CrossRef] [PubMed] [Google Scholar]
- Genotype-phenotype correlates of infantile-onset developmental & epileptic encephalopathy syndromes in South India: A single centre experience. Epilepsy Res. 2020;166:106398.
- [CrossRef] [PubMed] [Google Scholar]
- Sodium-channel defects in benign familial neonatal-infantile seizures. Lancet. 2002;360:851-2.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical spectrum of SCN2A mutations expanding to Ohtahara syndrome. Neurology. 2013;81:992-8.
- [CrossRef] [PubMed] [Google Scholar]
- De novo mutations of voltage-gated sodium channel alphaII gene SCN2A in intractable epilepsies. Neurology. 2009;73:1046-53.
- [CrossRef] [PubMed] [Google Scholar]
- A nonsense mutation of the sodium channel gene SCN2A in a patient with intractable epilepsy and mental decline. J Neurosci. 2004;24:2690-8.
- [CrossRef] [PubMed] [Google Scholar]
- SCN2A encephalopathy: A major cause of epilepsy of infancy with migrating focal seizures. Neurology. 2015;85:958-66.
- [CrossRef] [PubMed] [Google Scholar]
- The phenotypic spectrum of SCN2A-related epilepsy. Eur J Paediatr Neurol. 2020;24:117-22.
- [CrossRef] [PubMed] [Google Scholar]
- Mutations in the sodium channel gene SCN2A cause neonatal epilepsy with late-onset episodic ataxia. J Neurol. 2016;263:334-43.
- [CrossRef] [PubMed] [Google Scholar]
- Whole genome sequencing identifies SCN2A mutation in monozygotic twins with Ohtahara syndrome and unique neuropathologic findings. Epilepsia. 2013;54:e81-5.
- [CrossRef] [PubMed] [Google Scholar]
- SCN2A mutation in an infant with Ohtahara syndrome and neuroimaging findings: Expanding the phenotype of neuronal migration disorders. J Genet. 2019;98:54.
- [CrossRef] [PubMed] [Google Scholar]
- Oligodendrogenesis in the normal and pathological central nervous system. Front Neurosci. 2014;8:145.
- [CrossRef] [PubMed] [Google Scholar]
- Role of the sodium channel SCN9A in genetic epilepsy with febrile seizures plus and Dravet syndrome. Epilepsia. 2013;54:e122-6.
- [CrossRef] [PubMed] [Google Scholar]
- Pathogenic mechanism of recurrent mutations of SCN8A in epileptic encephalopathy. Ann Clin Transl Neurol. 2016;3:114-23.
- [CrossRef] [PubMed] [Google Scholar]
- De novo pathogenic SCN8A mutation identified by whole-genome sequencing of a family quartet affected by infantile epileptic encephalopathy and SUDEP. Am J Hum Genet. 2012;90:502-10.
- [CrossRef] [PubMed] [Google Scholar]
- The phenotypic spectrum of SCN8A encephalopathy. Neurology. 2015;84:480-9.
- [CrossRef] [PubMed] [Google Scholar]
- Voltage-gated sodium channel ß subunits and their related diseases. Handb Exp Pharmacol. 2018;246:423-50.
- [CrossRef] [PubMed] [Google Scholar]
- SCN1B-linked early infantile developmental and epileptic encephalopathy. Ann Clin Transl Neurol. 2019;6:2354-67.
- [CrossRef] [PubMed] [Google Scholar]




