
Translate this page into:
Mutation Location and Cognitive Impairment in Duchenne Muscular Dystrophy
Address for correspondence: Dr. Soumava Mukherjee, Department of Neurology, Nil Ratan Sircar Medical College and Hospital, 138 A.J.C BOSE ROAD, Kolkata - 700 014, West Bengal, India. E-mail: dr.soumava@gmail.com
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Background:
Cognitive impairment is commonly seen in patients with Duchenne muscular dystrophy (DMD). Few studies have shown a correlation between loss of different isoforms of the DMD gene and cognitive impairment.
Objective:
The objective of the study was to determine whether correlation exists in the location of mutation in DMD gene or loss of different isoforms and cognitive impairment in children with DMD in the Indian population.
Materials and Methods:
Ten children were evaluated. Gene mutation analysis was done by multiplex ligation-dependent probe amplification method. The isoforms affected were inferred from mutation location in each of these patients. Binet Kamat Intelligence Test (BKT) and Bender Gestalt test (BGT) were administered.
Results:
All male patients were aged between 4 and 9 years. Genetic analysis showed deletion in all patients, with seven having deletion in “hotspot” regions (exon 43–52). Psychometric analysis by BGT and BKT showed mean score of 8.6 and mean IQ score of 85.5, respectively. Comparison between patients with hotspot mutations and mutations in other regions, for mean IQ score and BGT score, was statistically significant (P = 0.132 and P = 0.005, respectively). The difference in the IQ score between patients with isolated Dp427 loss (n = 3) and cumulative Dp427/Dp260/Dp140utr loss (n = 6) was statistically significant (P = 0.011). Visuomotor functioning was more impaired in patients with isolated Dp427 loss.
Conclusion:
The role of cumulative loss of isoforms along with importance of loss of Dp140pc isoform was seen in our study. One patient with loss of Dp140utr isoform had intellectual impairment which is not commonly seen. Visuomotor functioning is more affected in more upstream mutations as shown in our study.
Keywords
Cognitive impairment
Duchenne muscular dystrophy
Mutation Location

INTRODUCTION
The prevalence of Duchenne muscular dystrophy (DMD) in the general population is approximately 3/100,000.[1] The inheritance is X-linked recessive, but almost one-third of the cases are sporadic, presumably due to a spontaneous mutation. Affected children are normal at birth, but during the 2nd year when the boys start walking, the clumsiness noticed in toddlers persists. The weakness gradually progresses, and the child becomes wheelchair bound by an average age of 12.2 years.[1]
The gene responsible for this dystrophy is located in Xp21 locus which comprises 79 exons or coding regions. Most of the mutations are due to deletions or duplications, but point mutations have also been observed. “Hot spots” for these gene deletions have been observed, notably between exons 43 and 52, with exons 44 and 49, particularly, have been affected.[1]
The neuropsychological examination in patients with DMD has shown a reduction in mean Full-Scale Intelligence Quotient (FSIQ) by approximately 1 standard deviation with respect to the population mean in various studies, with severity ranging from borderline to severe intellectual disability. About 19%–35% of DMD patients have been found to have FSIQ <70, with 3% having moderate-to-severe intellectual disability (FSIQ <50%).[2] Most studies have shown a greater impairment in verbal intelligence quotients than performance intelligence quotients.[2] A significant difference between the allelic disorders, DMD and Becker muscular dystrophy, has also been shown. Studies have failed to show a simple relationship between the degree of weakness and cognitive impairment in children with DMD which suggests that these two phenotypes may have a tissue-specific control.[2] The DMD locus produces many isoforms from different promoters, of which at least seven have been identified. Three full-length isoforms are derived from unique upstream exon sequences (Dp427c, m, and p). Downstream introns produce at least 4 shorter mRNA products (Dp260, Dp140, Dp116, and Dp71). These isoforms exhibit cell type specificity. Among these, isoforms Dp71 and Dp140 are abundant in fetal brain, which suggests that they may be of particular significance to the cognitive deficits. Initial studies showed that deletions involving exon 52 were more frequently associated with cognitive impairment, but later studies pointed to a more causal effect of different dystrophin isoforms.[23]
MATERIALS AND METHODS
Ten children admitted in our Department of Neurology in Nil Ratan Sircar Medical College in 2017 with a diagnosis of DMD were studied. All these male children presented with progressive weakness. The diagnosis of DMD was confirmed in all the patients, apart from routine investigations, with genetic analysis by multiplex ligation-dependent probe amplification method for exon deletion, and the exon deletion location in each of them was documented. The isoforms affected were inferred from mutation location in each of these patients. Detailed psychometric analysis was undertaken in all the patients. Visuomotor perceptual organizational skills and IQ scores were documented by Bender Gestalt Test (BGT) and Binet Kamat Intelligence test (BKT). BKT, which is standardized for the Indian population, is administered for patients aged between 3 and 22 years and evaluates several domains such as judgment, reasoning, memory, comprehension, word definition, and problem-solving. Informed consent was obtained from all the patients.
Statistical analysis involved parametric one-way t-test for comparison between IQ scores and raw scores of BGT between two groups of patients.
RESULTS
All the male patients were aged between 4 and 9 years, with a mean age of presentation at 7.3 years. Genetic analysis showed deletion in all the patients, with seven patients having deletion in “hot spot” regions (exons 43–52). Psychometric analysis by BGT showed a mean score of 8.6. IQ score by BKT showed a mean score of 85.5 [Table 1].
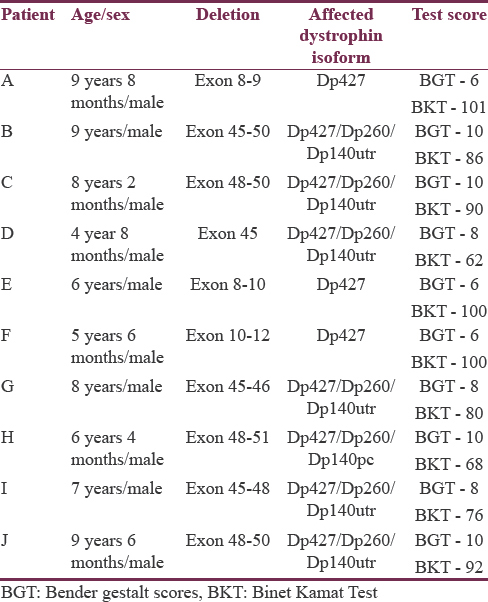
For the purpose of comparison, patients were divided into two groups. The first group had patients with a deletion in hot spot regions and the second group in other regions of the DMD gene. The mean IQ score was compared between these two groups, and the difference was significant (P = 0.132). The mean BGT scores between the two groups, when compared was also significant (P = 0.005).
Three patients had isolated Dp427 isoform loss, with a mean IQ score of 100.3. Six patients had loss of the Dp427/Dp260/Dp140utr isoform, with a mean IQ score of 81. The difference in the IQ scores between these two groups was statistically significant (P = 0.011) [Table 2]. One patient had Dp140pc isoform loss, and the FSIQ score was 68.

Visuomotor functioning (BGT raw scores) was more impaired in patients with loss of Dp427 isolated loss than in patients with cumulative loss of Dp427/Dp260/Dp140utr (P = 0.001) [Table 2].
DISCUSSION
The general intellectual level in our study group was lower than normative values. Similar results were shown by Milic Rasic et al.[3] Two children had intellectual impairment with an IQ <70 (20%) which is in agreement with other studies (19%–35%), though the number of children studied in our group was less.
All the children in our study had exon deletion in the DMD gene, so it was not possible to ascertain the effect of the type of mutation on the intellectual level. However, many studies have shown that this may not have a causal effect on the level of intelligence in these patients.[23]
Previous studies have found association between intellectual disability and location of mutation when ascertained between groups of children with mutations occurring upstream or downstream of exons 30 and 45.[2] Subsequent studies showed more correlation when mutation location in relation to their functional consequence and loss of expression of different dystrophin isoforms were considered. In our study, there was a significant difference in mean IQ scores between patients with mutations in the hot spot region and mutations in other regions of the DMD gene (P = 0.132). The visuomotor skills as depicted by the raw BGT scores were also significantly different between the two groups (P = 0.005).
The loss of Dp427 isoform is common in DMD children, and it has been postulated to be important in cognitive impairment.[3] This isoform is expressed in neocortex, cerebellum, and amygdala and has structural and functional (regulation of Gamma Aminobutyric acid a receptors) role. In our study, three patients had isolated loss of Dp427 isoform, but none had intellectual impairment (IQ <70). However, two patients with impairment (IQ <70) had mutations affecting Dp140 and Dp260 isoforms in addition to Dp427 which may point toward a role of cumulative loss of these isoforms. A similar finding was observed in the study by Milic Rasic et al.[3]
Previous studies have shown a statistically significant difference in the mean FSIQ when patients with mutations in the 5’ untranslated region (Dp140utr) and protein coding region (Dp140pc) were compared.[3] Mutations in the Dp140utr region have been found to have a lesser effect on FSIQ when compared to Dp140pc isoform loss.[23] In our study, one patient with Dp140pc isoform loss had intellectual impairment (IQ 68). However, one patient (16%) with Dp140utr isoform loss had a similar intellectual impairment (IQ 62).
The Dp71 isoform is abundantly expressed in fetal and adult brain, particularly in cortex and hippocampus.[3] Due to the low frequency of mutations in the most distal part of the DMD gene, this isoform loss is less common, but it has been found to have a profound effect on the intellectual ability.[2] No patient in our study had a loss of this isoform.
Furthermore, patients with isolated loss of Dp427 isoform had more affection of visuomotor functioning than patients with more downstream exon deletions.
In summary, this study conforms to hypothesis that loss of different dystrophin gene isoforms may have a more important role in determining intellectual ability than location of mutation alone. Furthermore, the cumulative loss of isoforms in affecting cognition was confirmed along with an important role for loss of Dp140pc isoform. One patient with loss of Dp140utr isoform had intellectual impairment which is not commonly seen. Visuomotor functioning is probably more affected in more upstream mutations as shown in our study. In addition, all the three children with isolated Dp427 isoform loss had evidence of autism-like behavior. Similar observations have been made by Milic Rasic et al.[3]
This study has certain limitations such as the small number of patients studied, but in a resource-poor country like ours, by studying the location of mutation and isoform analysis, we could provide the parents with information about the intellectual abilities of the child, which they are often more concerned about. This study is also probably the first undertaken to attempt to find a correlation between intellectual ability and mutation location in DMD patients in the eastern part of our country.
CONCLUSION
In conclusion, our study demonstrates the effect of cumulative loss of dystrophin gene isoforms in determining intellectual ability in children with DMD. Visuomotor functions are more affected by upstream muations. Loss of isolated Dp140utr isoform could have an impact on intellect, as was demonstrated in our study.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- Disorders of skeletal muscle. In: Bradley's Neurology in Clinical Practice (7th ed). Philadelphia, PA: Elsevier; 2016. p. :1921-23.
- [Google Scholar]
- Dystrophin gene mutation location and the risk of cognitive impairment in duchenne muscular dystrophy. PLoS One. 2010;5:e8803.
- [Google Scholar]
- Intellectual ability in the duchenne muscular dystrophy and dystrophin gene mutation location. Balkan J Med Genet. 2015;17:25-35.
- [Google Scholar]




