
Translate this page into:
Study of Clinical Features and Diagnosis Pattern of Duchene Muscular Dystrophy in Southern India
 , Anka Rao Areti1, S.N. Koteswara Rao G.1, Uma Sankar Kulandaivelu1, Rajasekhar Reddy Alavala1, Ravi Manne2
, Anka Rao Areti1, S.N. Koteswara Rao G.1, Uma Sankar Kulandaivelu1, Rajasekhar Reddy Alavala1, Ravi Manne2
This article was originally published by Thieme Medical and Scientific Publishers Pvt. Ltd. and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Abstract
Background Duchene muscular dystrophy (DMD) is an X-linked progressive muscle disorder that is characterized by proximal muscle weakness followed by a premature death in young boys. There is a low index of reports on diagnosis ratio and clinical features in Southern India.
Objective The present study aimed to conduct an observational survey on preliminary analysis, family history, associated complaints, and diagnosis ratio of DMD in southern regions of India.
Materials and Methods A systematic observation and survey were conducted on clinically confirmed DMD patients registered between 2019 and 2021 through the questionnaire. The questionnaire and pattern of study were identified by exploring published and unpublished studies available from electronic databases and critical assessment criteria considered by physicians. Preliminary analysis such as onset criteria, motor difficulties, milestone delay; family history and consanguinity analysis; chief complaints (ambulatory status, lordosis, respiratory, and cardiac outcomes), associated complaints such as enlarged tongue, oral hygiene, behavioral problems; and other similar parameters were studied. An assessment of the diagnosis rate and pattern was performed.
Statistical analysis The data were reviewed and interpreted through statistical methods mean ± standard deviation represented as a percentage.
Results In total, 400 DMD patients were included and 250 participated in the study. The onset age group was 2 to 5 years in 37% of the population. Milestone delay was seen in 86%; consanguinity marriage of parents was reported in 39%. Frequent falls were reported in 62% in 5 to 8 years old group. Wheelchair status was reported in 65% in 9 to 12 years old. Cervical and lumbar lordoses were seen in 57 and 69%, respectively, in above 13 years old. Respiratory and cardiac complications were 88 and 78% reported in above 13 years old, respectively. Other major associated complaints such as enlarged tongue were reported in 79%. Fifty-one percent underwent genetic diagnosis and 79% of the population underwent serum creatine phosphokinase (CPK) analysis for the confirmation of DMD.
Conclusion In this study population of South India, milestone delay was a major observation. Although there was a slight margin, family history shows “no blood relation among parents” in the majority of the study population. Chief complaints were predominantly severe above 13-year age group population. Serum CPK was the first choice for the first investigation, which is followed by a genetic diagnosis.
Keywords
muscle disorders
muscular dystrophies
outcome
muscle biopsy
mutation
consanguinity

Introduction
Muscular dystrophies are rare genetic disorders. Genetically, there are more than 50 types of muscle dystrophies and their subclasses are also involved based on the specific mutation. These are clinically identified by difficulty in walking, muscle degeneration, and muscle weakness. These clinical symptoms are variable depending on the specific mutated gene.
Duchene muscular dystrophy (DMD) is a heterogenous, X-linked genetic disease1 2 predominantly observed in young boys. Females act as non-symptomatic carriers. However, there are rare non-confirmed cases reported with mild symptoms. This progressive muscle disorder has no cure till date. The incidence of DMD occurs in a ratio of 1 in 3,000 to 5,500 male births3 and is caused due to the lack of “dystrophin” protein that affects muscle functionality. There are very few case studies reported; according to PubMed, 150 cases were reported4 till 2017. However, there is an increase in this margin since past few years, i.e., additional 126 case report studies were reported till date from 2017. In this study, we focused on the pattern of symptom identification and diagnostics reported in a group of 250 affected DMD populations. A detailed prospective study was conducted for a comparative analysis of DMD patients in different parts of Southern India with the aid of Amaravathi Muscular Dystrophy Association (Registration no: 33 of 2019). The study mainly focused on the symptoms, diagnosis pattern experienced by patients, and onset criteria of DMD.
This study focuses on the fusion of symptoms experienced and genetic diagnosis reports. Improvements can result from these kinds of studies in the margin of investigations and research depending on the patient outcomes. Case report analysis in Southern India is rare. This may be because of the dearth of knowledge on DMD. Therefore, there is a poor expected outcome in these patients. Diagnosis of DMD needs a comprehensive understanding of family history,5 medical history,6 age of onset, disease-specific features, onset, and development of distribution of weakness.
Muscle biopsy is the most practiced investigation for the diagnosis of DMD.7 Genetic diagnosis test is still a challenge. Even if the testing procedures are available due to price trends, it is a prohibitive factor to many low-income patients. One of the most reliable diagnostic tests of DMD is multiple polymerase chain reaction (mPCR). The mPCR consists of multiple primer sets within a single primer mixture. In this, predefined mutations are detected that is one of the greatest weaknesses in general. However, there is a need for extensive genetic studies to assess the unique mutations in the patients.8 The cost factors also play a crucial role in the hindrance of the research studies. But the importance of these tests is still high.
Materials and Methods
A survey9 was conducted through sharing a questionnaire link in the advocacy group, Amaravathi Muscular Dystrophy Association (Registration number: 33 of 2019) of patients registered between 2019 and 2021, and also through a detailed phone interview. Minor changes were made in the methodology adopted by Romitti et al.9 The questionnaire was developed based on the existing research work on DMD10 and assessment criteria considered by the physicians for drawing conclusion as reported. In addition, survey parameters were also considered. All the parameters of the questionnaire were detailed to patients and parents/guardians before the initiation of the study, and their consent was taken by explaining the details of the consent form. Parents were involved in reporting the details of young and debilitated children and described the ramifications that affected a DMD child.
The study population was divided into “groups.” The study population was divided into three groups for the assessment of chief complaints, but other associated parameters and diagnosis pattern were not interpreted through this method. The reason for dividing into groups based on the age for chief complaints was due to wild variance seen with age in the parameters such as motor difficulties, ambulatory factors, cardiac, and respiratory complaints. Diagnosis parameters and associated complaints did not show variance with the age factor. Hence, the statistics of these parameters were studied at the screening stage (not grouped). Patients of different age groups were included. Patients suffering from other muscle disease like multiple sclerosis and other types of muscular dystrophy were excluded. The data were reviewed and interpreted through statistical methods mean ± standard deviation represented as a percentage.
Results
DMD was mostly observed in boys, and the ideal identification age was early childhood. Milestone delay was seen in 86% of study size, 12% of the population reported no milestone delays, and 2% population reported unknown information (Table 1). Ideally, the age of onset was reported to be below 5 years, i.e., 2 to 5 years age group in 37% of the study. Age of onset was reported to be between 5 and 8 years age group in 33%, 9 and 12 years age group in 29%, and above or equal to 13 years in a trifling percentage, i.e., 1% of the study population (Table 2). Patients with a different dietary group did not show much variance in the postponement of the onset of symptoms; although 2% of the non-vegetarian diet population reported good management of symptoms, it is considered as negligible.
|
Parameter |
Percentage of population reported (%) |
|---|---|
|
Milestone delay |
66 |
|
No milestone delay |
32 |
|
Milestone delay unknown (not noticed) |
2 |
|
Percentage of population reported |
|||
|---|---|---|---|
|
Parameter assessed |
5–8 y |
9–12 y |
≥13 y |
|
Walk without support |
15 |
12 |
3 |
|
Walk with support |
55 |
23 |
8 |
|
Wheelchair bound |
30 |
65 |
89 |
|
Age of onset (37% reported <5 y) |
33% |
29 |
1 |
|
Cervical lordosis |
14 |
29 |
57 |
|
Lumbar lordosis |
3 |
28 |
69 |
|
Motor difficulties |
43 |
56 |
81 |
|
Frequency of falls |
28 |
62 |
9 (most of population is wheelchair bound) |
|
Gower's sign |
39 |
42 |
6 (most of population is wheelchair bound) |
|
Calf hypertrophy |
10 |
12 |
12 |
|
Muscle thinning, muscle twitching |
10 |
11 |
12 |
|
Cardiac issues |
19 |
25 |
78 |
|
Respiratory problems |
22 |
28 |
88 |
Family history was studied in the study groups. Consanguinity marriage of parents was reported in 39%, no blood relation of the affected parents was reported in 46%, far relation of parents was reported in 12%, and unknown relation was reported in 3% of the study population (Fig. 1). Thirty-three percent of the affected population reported that there is an immediate affected male sibling (mostly DMD), and only 1% reported affected female siblings whose genetic confirmation is not done (Fig. 1).

-
Fig. 1 Consanguinity analysis in study population.
Fig. 1 Consanguinity analysis in study population.
The study population was analyzed for the ambulatory condition as the chief parameter. At the age of 5 to 8 years, 15% of the population (Table 1) was able to walk without support, 55% of the population reported walking with support, and 30% of the population were wheelchair bound. In the age group of 9 to 12 years, 12% population reported the ability of walking without support, 23% of the population reported walking with support, and 65% of the population was wheelchair bound or partially used a wheelchair. The percentage of walking ability was gradually decreased or declined as the age progressed. The wheelchair bound status tremendously increased in the age group of 13 years and above to 89%; however, 8% reported walking with support and 3% reported walking without support (Table 2).
Cervical lordosis was reported in 14% of the population in the age group of 5 to 8 years, 29% in the age group of 9 to 12 years, and 57% in the age group of 13 years or above. Lumbar lordosis was reported in 3% of the population in the age group of 5 to 8 years, 28% in the age group of 9 to 12 years, and 69% in the age group of 13 years or above (Table 2).
Motor difficulties were reported in 43% in the age group of 5 to 8 years, 56% in the age group of 9 to 12 years, and 81% in the age group of 13 years or above. The frequency of falls was reported in 28% in the age group of 5 to 8 years, 62% in the age group of 9 to 12 years, and 9% in the age group of 13 years or above. Gower's sign was reported in 39% in the age group of 5 to 8 years, 42% in the age group of 9 to 12 years, and 6% in the age group of 13 years or above. Calf hypertrophy was reported in 10% in the age group of 5 to 8 years, 12% in the age group of 9 to 12 years, and 12% in the age group of 13 years or above. Muscle thinning and twitching were reported in 10% in the age group of 5 to 8 years, 11% in the age group of 9 to 12 years, and 12% in the age group of 13 years or above (Table 2).
The chief complaints of DMD patients were cardiac and respiratory difficulties that were reported in 19% of population in 5 to 8 years age group, 25% in 9 to 12 years age group, and 56% in 13 years or above age group (Table 2).
Associated complaint analysis was reported. Oral examinations were another important feature analyzed in the study population. Enlarged tongue was reported in 79%, left and right crossbites were reported in 45%, and poor oral hygiene was reported in 57% of the study population (Table 3). Posterior maximum voluntary force bite and labial force were reported in 77% population and unknown in 22% population. Speech and learning difficulties were reported in 45% of the population. Behavioral problems were reported in 57% of study population (Table 3).
|
Parameter |
Percentage of population reported (%) |
|---|---|
|
Enlarged tongue |
79 |
|
Left and right crossbites |
45 |
|
Poor oral hygiene |
57 |
|
Posterior maximum voluntary force bite |
77 |
|
Speech and learning difficulties |
45 |
|
Behavioral problems |
57 |
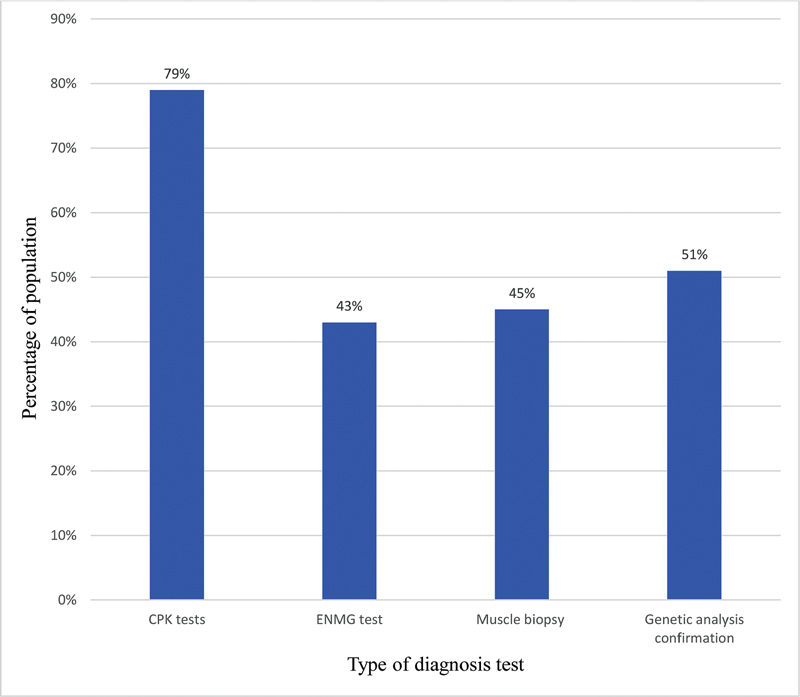
The study population variably reported that the primary diagnosis was through clinical observation and symptom identification. This is primarily confirmed by serological analysis (Table 4). The study population was analyzed for the pattern of diagnosis undergone. Seventy-nine percent of the study population reported undergoing creatine phosphokinase (CPK) tests, 43% electromyography (EMG) test, 45% muscle biopsy, and 51% genetic analysis confirmation (Fig. 2). Serological analysis revealed elevation of creatine kinase levels to 5,000 to 8,000 U/L (69% study population reported) and >300 ≤ 5,000 U/L in 31% of population. The elevations are reported for both alanine transaminase levels and lactate dehydrogenase levels that are reported as between 120 to 300 U/L and 400 to 700 μg/dL, respectively, in all the age groups of the study population. However, the 79% population reported further examination through electromyographic pattern and tissue invasive tests. Out of this 79% population, 55% population reported final genetic confirmations of mutation changes. It was reported that the physicians did not recommend undergoing any genetic confirmation of mutation changes.
-
Fig. 2 Diagnosis report in study population.
Fig. 2 Diagnosis report in study population.
|
Chief examinations |
Examination of muscle problems such as muscle weakness, degeneration, and tonicity |
|
Serological examinations |
Creatinine kinase enzyme analysis |
|
Associated complaint analysis |
Respiratory, cardiac, and oral examinations |
|
Muscle biopsy |
Primary muscle test for dystrophy confirmation |
|
Genetic analysis |
Tested for mutations and deletions of gene |
Discussion
There are many studies on DMD with irregular and improper reporting. This observational survey study focused on systematic and in-depth analysis of the pattern of identification and diagnosis of 400 DMD population, 250 patients accepted the consent to participate in the study. DMD is rare and pernicious among all types of dystrophies.11 DMD gene consists of 79 exons and a large number of introns. Because of the presence of a huge number of introns, these are prone to mutations.12
The affected population of DMD is mostly boys and the starting age is between 2 and 5 years. Predominantly, the age of onset observed is in early childhood with initial symptoms of walking difficulties. Overall, the reported dietary group (high proteinaceous and less proteinaceous diet) suggests that there is no major effect of the diet on the onset age of the clinical symptoms. But the symptomatic management was reported with adequate intake of proteins, which was acknowledged by a smaller group of patients in a negligible ratio. Since the progression risk is very high in the DMD population, the effect of protein and non-protein diet is less known and unidentified by patient groups or patients.
According to studies, consanguinity is the notion in 88% of the study population.13 Consanguinity is widely practiced in Asian countries especially in India. DMD is X-linked inherited where consanguinity is not an entailed factor. However, most of the patients reported that consulted physicians mostly attributed DMD to consanguine marriages. However, reports suggested “consanguinity” is not the key factor for DMD. Also, the study reported that the occurrence of DMD was seen in the immediate siblings of the DMD child.14 The symptoms, diagnosis, onset, and cessation of ambulation are assessed based on the symptoms of the older siblings. Due to this imaginary assessment, a negligible number of young female siblings were also reported without any genetic confirmation or primary diagnostic pattern. This imaginary confirmation is the primary factor of misconception that affects the management therapy and clinical trial enrollment.
Chief examinations include general physical assessments. Affected boys clinically reported ambulatory difficulty. Walking difficulty is severe in the above 13 years age group, while it initially starts in the group between 5 and 8 years of age. This can be attributed to increased progression of muscle weakness. Milestone delays such as walking were the key factor observed in infants and early childhood, and this factor is responsible for the primary identification of muscle disease. Cervical lordosis was merely increased in the above 12 years age group. Cervical and lumbar lordoses were mild in between 5 and 10 years age group and worse in above 12 years of age. Motor difficulties such as difficulty in rising from floor and frequent falls were estimated by the scale which is constructed based on the observational data of the study. Difficulties ought to be severe in the age group of 9 to 12 years and above. During the initial stages, affected boys used arm support to rise from the sitting position, which can be considered as “Gower's sign.” This is attributed to low strength of hip and thigh muscles. As age increases, the deterioration of muscles progresses, thereby degenerating muscle movement. There were similar scenarios reported with parameters such as calf hypertrophy, muscle tone, muscle thinning, and twitching. Cardiac and respiratory difficulties were majorly reported. The age group above 13 years encounters a high risk of cardiac failures. This fact is also suggested by the case report study.15 The weak peripheral and respiratory muscles occur in parallel that leads to respiratory and heart failure.
Oral manifestations include left and right crossbites, poor oral hygiene, voluntary force bite, and labial force. These can be attributed to orofacial muscle involvement that is apparently caused by the disease. Other problems reported predominantly include behavioral problems, speech, and learning difficulties. Behavioral problems may be attributed to psychological stress and anxiety. Normally, a child should be able to speak between the age of 18 to 24 months. The words may be mumbled. Also, the same scenario can be applied in the case of learning parameters such as walking, standing on own foot, and other motor functions. Here, in this study, we have reported difficulties in learning, speech, and behavioral parameters by considering non-developmental symptoms till 24 months of age.
As reported, the primary diagnosis is done by clinical assessment, findings, and correlation of the results. These findings were confirmed by serum creatinine levels and histopathology tests such as muscle biopsy and immune pathology.16 Further, it is genetically confirmed to understand the deletions and mutations in the gene. Leakage of proteins into serum is observed due to repeated contractions and increased permeability of sarcolemma. Due to this, creatinine levels are elevated and help in primary diagnostic confirmation of any muscle damage17 (Table 4).
Secondary dystrophy confirmation is through electroneuromyography and muscle biopsies. Electromyography involves the measurement of muscle response with respect to the nerve stimulation of that muscle. This test involves small electrodes that are inserted through the skin into the muscle.
A biopsy involves making a small cut in the skin near the thigh area. The muscle tissue is examined and studied for the identification of the degeneration, regeneration of muscle fibers, and variation in fiber size.13 These techniques cause irreversible changes, and these inclusive procedures bothered and were unacceptable by the patient groups in terms of short-term pain and long-term scars.
The study population in this study majorly reported undergoing serum and creatinine tests (serological examinations). Secondary confirmation reported was through invasive tests. Out of the population who underwent the invasive testing procedure, only 55% of the population reported undergoing genetic diagnosis. This is considered as the stumbling block of clinical trials and research advancements. The genetic diagnostic techniques aim at studying deletions and mutations that act as the most important criteria for mutation-specific therapies that are under development.18 According to recent studies, muscle biopsies are optional since the biopsies identify the DMD phenotype but not the mutations in the DMD gene.
Conclusion
This study bestows the affected DMD study population. Critical care should shadow children between 10 and 13 years and above age group, as there is a high risk of muscle weakness involved and also cardiac and respiratory muscle malfunction. In this study population of South India, milestone delay was the major observation. Although there was a slight margin, family history shows “no blood relation among parents” in the majority of the study population. Chief complaints were predominantly severe in the age group population of 13 years and above. Serum CPK was the first choice for the first investigation that is confirmed by genetic diagnosis.
Risk-benefit analysis studies revealed that the patients with no genetic confirmation were prone to high risks. Parents can consider family planning by knowing the hereditary pattern and confiscating suitable measures in planning the next child to terminate the hereditary occurrence of the disease. Also, there are suitable guidelines for the management of specific mutations and this leads to the probability of undertaking available therapy.
The progression of muscle weakness is irreversible, and hence, it is important to identify the disease at the earliest and follow available supportive therapies and management. The clinical consultants and primary physicians should inculcate complete diagnostic procedures to avail patients of the emerging gene therapies.
Conflict of Interest
None declared.
References
- A pilot survey study of adherence to care considerations for Duchenne muscular dystrophy. PLoS Curr. 2018;10:10.
- [Google Scholar]
- Duchenne muscular dystrophy: case report and review. J Family Med Prim Care. 2017;6(3):654-656.
- [Google Scholar]
- Dystrophin gene analysis in Hungarian Duchenne/Becker muscular dystrophy families—detection of carrier status in symptomatic and asymptomatic female relatives. Neuromuscul Disord. 2009;19(2):108-112.
- [Google Scholar]
- Muscle biopsies in clinical trials for Duchenne muscular dystrophy—patients' and caregivers' perspective. Neuromuscul Disord. 2019;29(8):576-584.
- [Google Scholar]
- From gene to disease; the dystrophin gene involved in Duchenne and Becker muscular dystrophy. Ned Tijdschr Geneeskd. 2002;146(8):364-367.
- [Google Scholar]
- Prevalence of Duchenne and Becker muscular dystrophies in the United States. Pediatrics. 2015;135(3):513-521.
- [Google Scholar]
- The clinical spectrum of limb girdle muscular dystrophy. A survey in The Netherlands. Brain. 1996;119:1471-1480. (Pt 5):
- [Google Scholar]
- Diagnostic accuracy of phenotype classification in Duchenne and Becker muscular dystrophy using medical record data1. J Neuromuscul Dis. 2018;5(4):481-495.
- [Google Scholar]
- Duchenne muscular dystrophy: a clinical, histopathological and genetic study at a neurology tertiary care center in Southern India. Neurol India. 2009;57(6):734-738.
- [Google Scholar]
- A rare case report of neurodegenerative disease: Duchenne muscular dystrophy in two male siblings. Int J Clin Pediatr Dent. 2015;8(2):163-165.
- [Google Scholar]
- Muscle and cardiac therapeutic strategies for Duchenne muscular dystrophy: past, present, and future. Pharmacol Rep. 2020;72(5):1227-1263.
- [Google Scholar]
- Natural history of serum enzyme levels in Duchenne muscular dystrophy and implications for clinical practice. Am J Phys Med Rehabil. 2020;99(12):1121-1128.
- [Google Scholar]
- Duchenne muscular dystrophy and dystrophin: pathogenesis and opportunities for treatment. EMBO Rep. 2004;5(9):872-876.
- [Google Scholar]
- Genetic and clinical profile of patients of Duchenne muscular dystrophy: experience from a tertiary care center in Eastern India. Indian Pediatr. 2015;52(6):481-484.
- [Google Scholar]




