
Translate this page into:
Neuroacanthocytosis: A rare movement disorder with magnetic resonance imaging
Address for correspondence: Dr. MA Hashmi, MRI Section, EKO CT and MRI Scan Centre, Medical College and Hospitals Campus, 88-College St., Kolkata - 700 073, India. Email: ahashmidrrad@yahoo.co.in
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Medknow Publications and was migrated to Scientific Scholar after the change of Publisher.
Sir,
A 32-year-old young male of average build presented to us with tremors in both upper and lower limbs, intentional and resting, with choreo-athetotic movement in upper limbs and tone rigidity in both the limbs.Power was grade 4/5 in all the limbs. The patient had one episode of seizure. Magnetic resonance imaging (MRI) showed bilateral, almost symmetrical, hyperintense signal changes in anterior medial globus pallidus with surrounding hypointensity in the globus pallidus on T2-weighted images. These imaging features have been termed the “eye-of-the-tiger” sign [Figures1 and 2]. The patient's cerebro-spinal fluid revealed no gross abnormalities. His 24-hour urine copper was 3.10 µg/L (normal range2–30 µg/L) and his serum ceruloplasmin was 31.93 mg/100 ml (normal range 20–60 mg/100 ml). His peripheral blood smear showed plenty of acanthocytes [Figure 3]. His apolipoprotein A level was 109 (normal 104–202) and apolipoprotein B level was 121 (normal 66–133). On the basis of clinical findings, typical type of MRI images and large number of acanthocytes in peripheral blood, diagnosis of neuroacanthocytosis (NA) was made.
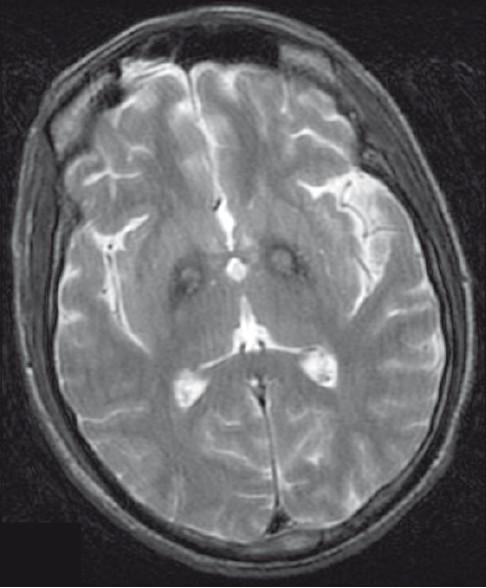
- T2-weighted image is showing symmetrical hyperintense signal changes in anterior medial globus pallidus with surrounding hypointensity in the globus pallidus. These imaging features have been termed the “eye-of-the-tiger” sign
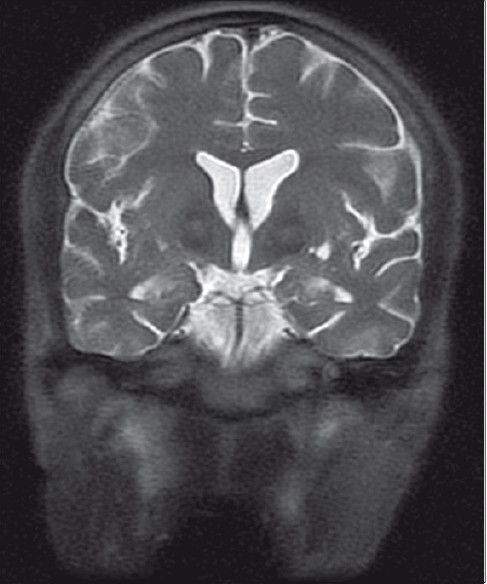
- Coronal T2-weighted images showing features as described in Figure 1
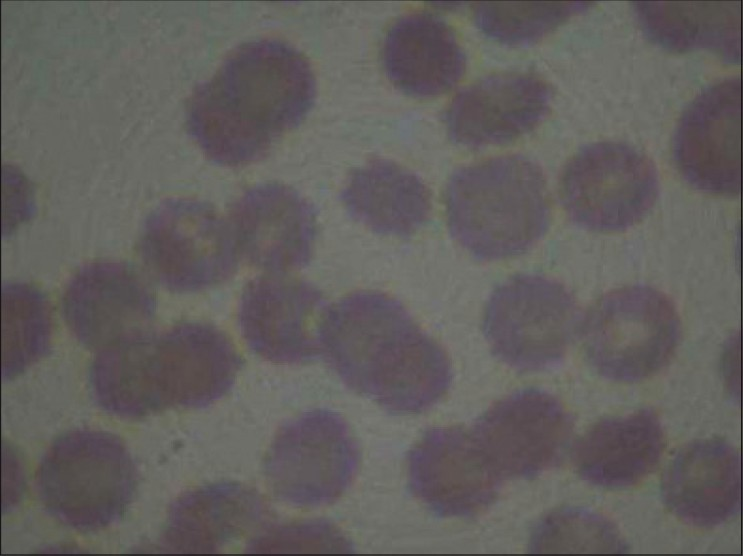
- peripheral blood smear stained with haemotoxylin is showing plenty of acanthocytes
NA syndromes include combined features of acanthocytosis,[1] i.e., spiked red blood cells, chorea, orofacial tics, amyotrophy and normobetalipoproteinemia. NA has been described to be inherited as an autosomal recessive disorder, as an autosomal dominant disorder, and as part of an X-linked disorder called McLeod syndrome (MLS). The first form of NA was described as Bassen-Kornzweig disease, or abetalipoproteinemia, which is an autosomal recessive abnormality of lipoprotein metabolism, resulting in ataxia combined with acanthocytosis. Bassen-Kornzweig disease was earlier compared with Friedreich ataxia. The two are rather similar except that patients with Bassen-Kornzweig disease have acanthocytes. Critchley described the second type of NA in 1960 and it was described by Levine later in 1968. The Levine–Critchley syndrome, as it came to be called, resembles Huntington disease (HD) with prominent choreiform or choreoathetoid movements, progressive dementia, and, in the original descriptions, autosomal dominant inheritance. One notable difference from HD is that Levine–Critchley syndrome manifests acanthocytes. Chorea-acanthocytosis, MLS, and HD can have movement disorders and acanthocytes. Neurodegenerative disorder is usually inherited as an autosomal recessive trait linked to chromosome 9q21.[2]
Severe involvement of caudate and putamen is seen with less severe changes seen in the pallidum.[34] Neuronal loss and mild gliosis can be seen in the thalamus, substantia nigra, and anterior horn of the spinal cord. Acanthocytes are seen in peripheral blood smears. In NA that does not involve abetalipoproteinemia or hypobetalipoproteinemia, lipoprotein levels are normal. Wilson's disease has typical MRI imaging[5] and there is a change in ceruloplasmin level. Hallevorden-spatz disorder can have MRI findings like that of NA but acanthocytes are not found in blood. HD can show atrophy of caudate. The hyperintensity in NA represents pathologic changes including gliosis, demyelination, neuronal loss, and axonal swelling, and the surrounding hypointensity is due to loss of signal secondary to iron deposition.
In betalipoprotein deficiencies, lack of microsomal triglyceride transfer protein (MTP) or a direct mutation in the gene for betapolipoprotein leads to a decreased absorption of vitamin E as well as other vitamins and possibly other cofactors. This leads to damage to the dorsal root ganglia, spinocerebellar tracts, retina, and cerebellum. It also leads to a defect in the conformation and/or fluidity of the erythrocyte membrane. Band three of the membrane appears to be one of the components significantly involved.
The case discussed here gives us an important message, i.e., typical MRI imaging finding “eye-of-the-tiger” sign appearance with associated peripheral blood smear study showing acanthocytes is useful in detecting this rare neurological disease.
References
- Clinical features and molecular bases of neuroacanthocytosis. J Mol Med. 2002;80:475-91.
- [Google Scholar]
- Developments in neuroacanthocytosis: Expanding the spectrum of choreatic syndromes. Mov Disord. 2006;21:1794-805.
- [Google Scholar]
- Follow-up magnetic resonance imaging in Hallervorden-Spatz disease. Clin Neurol Neurosurg. 1992;94:57-60.
- [Google Scholar]
- Subcortical white matter abnormalities related to drug resistance in Wilson disease. Neurology. 2006;67:878-80.
- [Google Scholar]




