
Translate this page into:
Strengthening molecular genetics and training in craniosynostosis: The need of the hour
Address for correspondence: Dr. Mayadhar Barik, Department of Pediatric Surgery, All India Institute of Medical Sciences, New Delhi - 110 029, India. E-mail: mayadharbarik@gmail.com
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Craniosynostosis (CS) is premature fusion of skull. It is divided into two groups: Syndromic craniosynostosis (SCS) and non-syndromic craniosynostosis (NSC). Its incidence in Indian population is 1:1000 live births where as in the USA it is 1:2500 live births. Its incidence varies from country to country. Molecular genetics having great interest and relevance in medical students, faculty, scientist, pediatric neurosurgeon and staff nurses, our objective was to educate the medical students, residents, researchers, clinicians, pediatric neurosurgeon, anesthetists, pediatricians, staff nurses and paramedics. We summarized here including with diagnosis, investigations, surgical therapy, induction therapy, and molecular therapy. Molecular genetics training is needed to know the information regarding development of skull, cranial connective tissue, craniofacial dysplasia, frame work, network of receptors and its etiopathogenesis. The important part is clinically with molecular therapy (MT) how to manage CS in rural sector and metropolitan cities need a special attention.
Keywords
Craniosynostosis
Indian population
medical education
training

Introduction
Current treatment of craniosynostosis (CS) consists of surgical modalities exclusively. Management of CS does not have to involve surgery, though surgery is an important modality. The patients are best managed in a multi-disciplinary way that should involve craniofacial surgeons, ENT surgeons, pediatricians, craniofacial nurse specialists and allied medical health professionals such as speech and language therapists and dieticians. Various surgical approaches have evolved to excise the prematurely fused sutures and to remodel the dysmorphic skull by cranial vaultre-modeling and advancement procedures. Apart from strip craniotomies of the affected sutures, the procedure involves extensive osteotomies and repositioning of bony plates to remodel the cranial vault. The goal of surgery is to increase intracranial volume (ICV) and to prevent elevation of intracranial pressure (ICP). As a result the surgical correction of CS is best begun within the first 3 to 6 months of life. Delay in correcting CS can exacerbate associated facial skeletal abnormalities such as facial asymmetry, malocclusion and strabismus. The exact surgery required depends on the sutures involved. For example, metopic synostosis is marked by a restriction in the volume of the anterior cranial fossa. This is treated by performing a sagittal advancement of the fronto-orbital bar, with particular focus on the lateral regions.[1] Midface hypoplasia and other craniofacial dysmorphisms often accompany syndromic forms of CS and are usually addressed at 4-5 years of age.[2] Despite advances made in craniofacial surgery for the treatment of CS, the operations are not without risk. These surgeries are usually performed in infancy, a time when children are most susceptible to physiological insults. Infection, optic nerve ischemia, seizures, bleeding, and the need for blood transfusion are significant events in infancy and early childhood.[3] We estimated that 98% of patients undergoing fronto-orbital advancement (FOH) procedures, 100% going cranial vault reconstruction (CVR), 32% spring cranioplasty findings hold a great challenge and new promise for anesthetists.[4] Surgical correction of CS often requires massive blood transfusions (MBTs).[5] MBTs can be defined as more than 10 units of packed red blood cells (RBCs) transfusion in 24 hours or replacement of more than 50% of patient blood volume within 12 to 24 hours.[6] Transfusion requirement exists in both syndromic and non-syndromic cases. The major part is hemorrhage, required special attention, surgical procedure and pre-operative vigilance.[7] This is compounded by the need for further surgery either for suture re-fusion after strip craniotomy or for correction of secondary defects. Mortality rates have been described to be as high as 1.5-2%.[8] For these reasons, the development of minimally invasive, biological based-therapies such as micro-injection, gene therapy (GT) offer a very attractive option for the treatment of CS.[9]
Background
According to our expertization and research, we formulate an innovative conceptual role model for CS management [Figure 1].
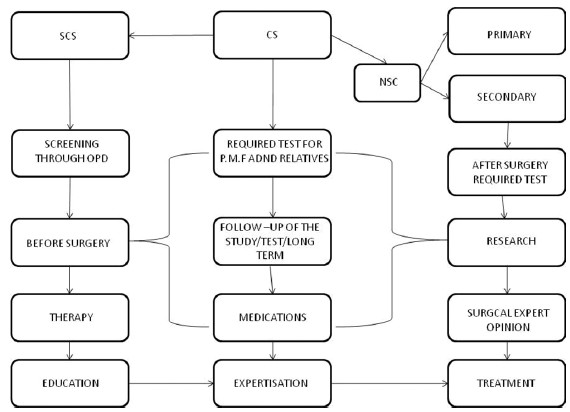
- An innovative conceptual model for craniosynostosis (CS) management
Investigations
(1) Skull radiograph, (2) 3D CAT SCAN, (3) funduscopy examination, (4) psychological tests, (5) molecular tests, (6) clinical photographs, (7) family history, (8) essential additional information from second degree relatives of father and mother, (9) measurement of skull diagram, (10) anthropometric analysis, (11) dental evaluation, (12) IQ test, and (13) coginitive, speech test and (14) behavioral analysis should be done.
Antenatal diagnosis
Prenatal testing is available for high risk pregnancies if a molecular defect has been identified in the family (such as an earlier affected child or an affected parent). Fetal DNA obtained through chorionic villous sampling (CVS) at about 10-12 weeks of gestation (preferably) or amniocentesis at 16-18 weeks of gestation can be analyzed for the known disease causing mutation. In a pregnancy not previously identified to be at risk for CS in which an abnormal skull shape is detected on prenatal ultrasound examination, prenatal testing is often not possible, unless other findings indicate a well-known syndrome form such as the limb anomalies in Apert syndrome.
Low risk craniosynostosis
A vast majority of patients do not have neurologic complications or mental retardation. Most patients present with either cosmetic deformity or sociopsychological problems.[7]
Intermediate risk craniosynostosis
This group of CS may be treated surgically by removing the affected sutures. These are complex procedures that require extensive planning and CT scan with three-dimensional reconstruction and a team of physicians including neurosurgeons and anesthesiologists. Correction is usually performed in the first year of life for intermediate risk CS patients.[8]
High risk craniosynostosis
Untreated CS at time of diagnosis usually require extensive investigation including are X-ray skull, funduscopy, size of skull, SPECT/CT or the cognitive and language development and IQ tests. There may be a greater developmental risk for infants with a single suture CS then? Those secondary to (1) Primary microcephaly, (2) hydrocephalus, (3) postural plagiocephaly. Recently we observed that advanced paternal age and higher parental education level are also associated with CS.[9]
Surgical therapy
No medical treatment exists to stop early ossification of a cranial suture. Reasons to perform surgical interventions can be increased intracranial pressure (ICP), decrease of skull growth below third centile, facial deformity (which can give cosmetic reasons for interventions), and (rarely) progressive exophthalmoses threatening the eyes. Not all children with a CS need surgical interventions. The results are best when surgery is performed between the age of 4 and 8 months at an early stage of although there may be reasons to postpone surgery till a later age.[10] Infants with a CS may require a series of surgical procedures.[1112] Procedures for correction of CS are performed in young infants with a small blood volume (SBV) yet represent major surgery with extensive blood loss (EBL).[1314]
Induction therapy
Pre-treatment with erythropoietin and iron combined with acute preoperative normovolaemic hemodilution (APNH) could decrease homologous blood transfusion during surgery in CS patients.[15] Ringers lactate (RL) solutions and normal (0.9%) saline (NS) are also useful because the surgery requires large amount of intravenous fluid replacement, which alter the ionic composition of body compartments.[16]
Molecular therapy
Bone morphometric proteins (BMP) are part of the transforming growth factor β (TGF-β) super family and are originally found to induce bone and cartilage formation.[17] This large group of proteins, comprising nearly one third of the TGF-β superfamily, has also been found to be involved in mesoderm induction, skeletal patterning, and limb development.[18] Immunohistological analysis of embryonic mouse sagittal sutures revealed expression of BMP-2 and BMP-4 at the osteogenic fronts, and BMP-4 in suture mesenchyme and osteogenic fronts of both fusing and patent sutures.[19] Novel tissue engineering techniques may allow the design of targeted complementary therapies to decrease the complications inherent in high-risk surgical procedures. Recent development in this filed include identifying genetic mutations, formulating etiopathogenesis of various craniosynostotic conditions, understanding cranial suture biology and molecular biochemical pathways involved in suture fusion and the design, development and application of various vehicles and tissue engineered constructs to deliver cytokines and genes to cranial sutures. (Many of genes involved in syndromic forms of (CS are shown in Table 1).
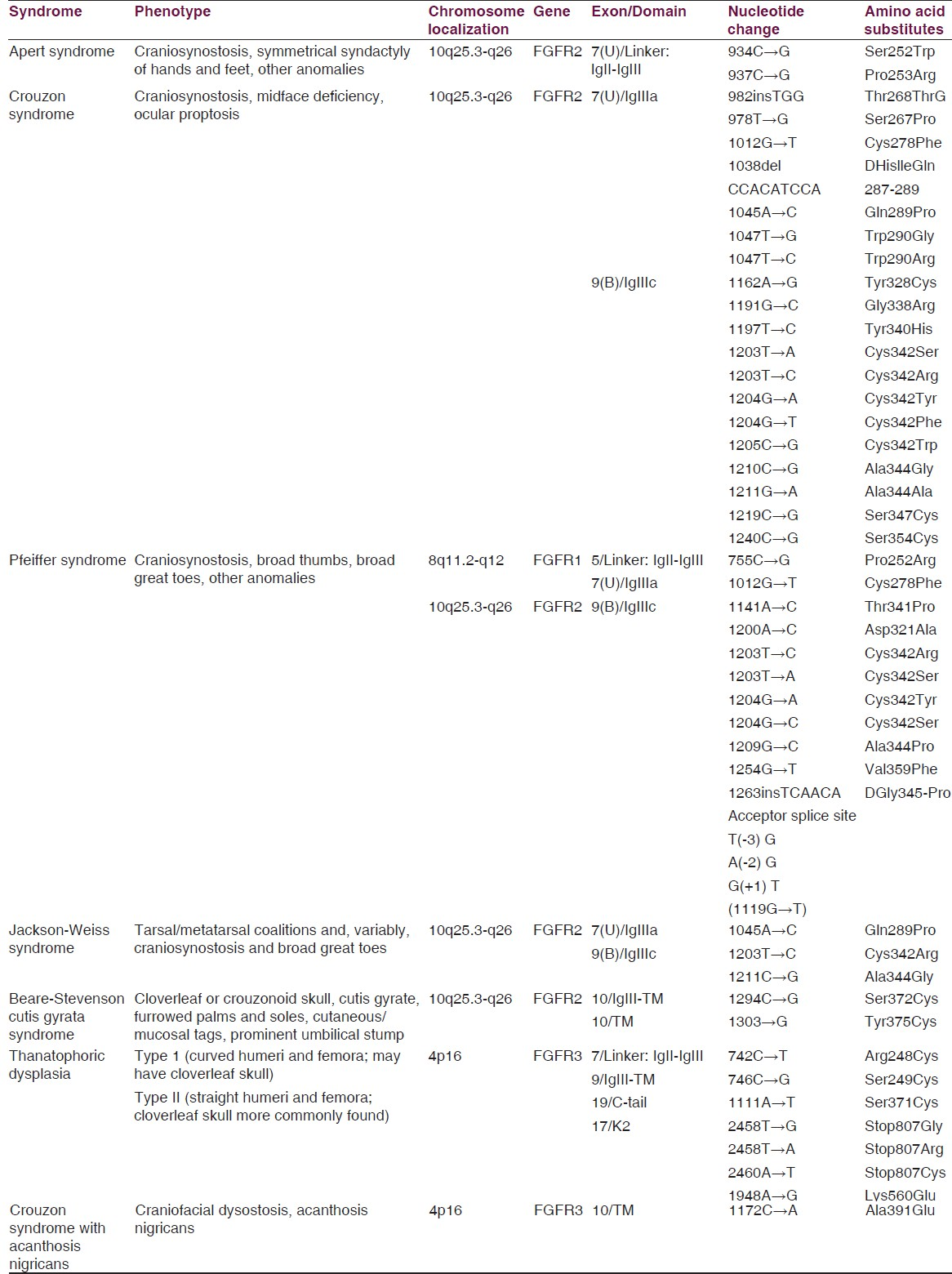
We find out novel and innovative mutation including with non-syndromic craniosynostosis (NSC), FGFR2iiia, FGFR2iiib, FGFRiiic has been given 80% (120). However, FGFR1, FGFR3 has observed lesser correlation with NSC. Hence, we found that FGFR2 gene and its isomers FGFR2iiia, FGFR2iiic genes are helpful for prognostic marker and 99m Tc ECD SPECT/CT test as a diagnostic marker as the gold standard for CS. Hence, biologically based therapies maybe used as surgical adjuncts to rescue fusing sutures or help to manage postoperative resynostosis.[20]
Genetic counseling
For adequate genetic counseling, it is important to make a distinction between isolated and syndromic forms of CS. Adequate evaluation for additional symptoms has to be performed in each child with CS. Autosomal dominant inheritance is the most common inheritance pattern of most syndromic forms of CS.[1920] The percentage of patients showing spontaneous new mutation is high. However, germ line mosaicism is also known to occur and should be discussed with the family. Human genetics is now at a critical juncture. The molecular methods used successfully to identify the genes underlying rare Mendelian principle are failing to find the numerous genes causing more common, familial non-Mendelian disease. The human genome project opened new avenues to identify unknown biological process in human disease like CS. This massive and multifactorial data generated by the present high-through put omics assays and explore the utility of system biology approaches in this subject.[21] New opportunities are being presented for unraveling the complex genetic basis at non-Mendelian disorders based on large-scale genome-wide studies.[22] Usually non-Mendelian pattern of inheritance can be identified in non-syndromic forms of CS, although rarely the isolated CS is familial, following an autosomal dominant pattern of inheritance with reduced penetrance. The recurrence risk in such cases is dependent on the nature of the suture involved.
Future Directions
Paralleling recent work on cranial suture biology has been the explosion of nucleic acid-based therapeutics and the delivery mechanisms of these biological products, while only two DNA-based drugs, fomivirsen, an antisense oligonucleotide for treatment of CMV retinitis, and Medicine, a p53 adenovirus for oncological applications, have been approved for use; countless other formulations are in various stages of clinical trials. Progress has been impeded predominantly by development of safe and effective techniques.[23] Nevertheless, medicine sits on the cusp of an exciting revolution where diseases are targeted at the genomic level. The application of these developments to CS presents exciting options to surgical intervention. As roles of FGF, TGF- TGF-b, EMX1, EMX2, MSX1, MSX2, TWIST, RECQL, BMP, BMP antagonists, and their respective receptors are being clarified in cranial suture biology, it is foreseeable that these molecules will be targets of therapy for CS. With an increased use of microarray technology, other potential candidate genes will also be identified. The goal would be to suppress the expression of genes promoting suture fusion or to increase expression of those which support suture patency. While the direct application of exogenous growth factors or neutralizing antibodies is an option, shortfalls of such approaches include short half-life, low bioavailability, enzymatic inactivation and high cost of purification. With such considerations, gene therapy holds great promise for the future of CS.[2024]
Conclusion
Before stopping my ink we have to think that as we move into a new millennium, the association of computational and molecular technological developments including the sequencing of the human genome, next generation sequencing is opening up new and unprecedented opportunities for genetics research. The important part is to devise a strategy to manage craniosynostosis (CS) in rural sector and metropolitan cities and training for medical student, researchers, clinicians, pediatric neurosurgeon and staff nurses is necessary.[2526272829]
Acknowledgment
We are thankful to ICMR (Indian Council of Medical Research) for funding us on due time.
Source of Support: Nil.
Conflict of Interest: None declared.
References
- Combined report of problems and complications in 793 craniofacial operations. Plast Reconstr Surg. 1979;64:198-203.
- [Google Scholar]
- Plastic Surgery (4th ed). Boston: Little, Brown; 1991.
- Anesthesia for surgery related to craniosynostosis: A review. Part 2. Pediatr Anaesth. 2013;23:22-7.
- [Google Scholar]
- Blood loss and replacement for paediatric cranioplasty in Australlia-a prospective national audit. Anesth Intensive Care. 2012;40:107-13.
- [Google Scholar]
- Blood-conservation techniques in craniofacial surgery. Ann Plast Surg. 2005;54:525-9.
- [Google Scholar]
- Craniofacial surgery. In: Evers BM, Townsend CM, Mattox KL, Beauchamp RD, eds. Pediatric Plastic Surgery (19th ed). Sabiston Text Book of Surgery: Elsevier Saunders; 2012. p. :1923.
- [Google Scholar]
- Craniosynostosis: Current treatment and future therapy. Gene Ther Mol Biol. 2005;19:423-30.
- [Google Scholar]
- Potential therapeutic strategies and its application in correcting birth defects, craniosynostosis, neurological disorders and other diseases. J Metabolic Synd. 2013;2:122.
- [Google Scholar]
- New trends in the 3D management of CT data in plastic and reconstructive surgery. Int Surg. 1997;84:332-8.
- [Google Scholar]
- Perioperative blood salvage during surgical correction of craniosynostosis in infants. Br J Anaesth. 2000;85:550-5.
- [Google Scholar]
- Craniosynostosis: Surgical treatment during the first year of life. J Neuro Sci. 1992;36:129-37.
- [Google Scholar]
- Craniosynostosis: An analysis of the timing, treatment, and complications in 164 consecutive patients. Plast Reconstr Surg. 1987;80:195-212.
- [Google Scholar]
- Timing of treatment for craniosynostosis and facio-craniosynostosis: A 20-year experience. Br J Plast Surg. 1994;47:211-22.
- [Google Scholar]
- Monitoring visual function in children with syndromic craniosynostosis: A comparison of 3 methods. Arch Ophthalmol. 2006;124:1119-26.
- [Google Scholar]
- Craniosynostosis: An assessment of blood loss and transfusion practices. Can J Anaesth. 1989;36:473-7.
- [Google Scholar]
- Non-invasive aortic blood flow measurement in infants during repair of craniosynostosis. Br J Anaesth. 1998;81:696-701.
- [Google Scholar]
- To elucidate the genotype-phenotype relationship in non-syndromic craniosynostosis by analysing the mutations of the FGFR1 and FGFR2gene with nuclear imaging test. Scientigraphy Non-oncology. Indian J Nucl Med. 2013;28:S16-20.
- [Google Scholar]
- Overexpression of BMP-2 and BMP-4 alters the size and shape of developing skeletal elements in the chick limb. Mech Dev. 1996;57:145-57.
- [Google Scholar]
- FGF-, BMP and Shh-mediated signaling pathways in the regulation of cranial suture morphogenesis and clavarial bone development. Development. 1998;125:1241-51.
- [Google Scholar]
- Genetic study of nonsyndromic coronal craniosynostosis. Am J Med Genet. 1995;55:500-4.
- [Google Scholar]




