
Translate this page into:
Mesenchymal Chondrosarcoma of the Brain with Metastasis: A Case Report with Literature Review
Ashish Sharma, MBBS, MS, MCh Department of Neurosurgery, SMS Hospital Jaipur, Rajasthan 302004 India drashuneuro@gmail.com
This article was originally published by Thieme Medical and Scientific Publishers Private Ltd. and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Abstract
Mesenchymal chondrosarcoma of the brain is one of the rarest tumors with dismal prognosis. A 26-year-old man presented with headache, vomiting, and diplopia. On evaluation, a moderately enhancing extra-axial lesion was seen in right temporal region involving right greater wing of sphenoid, adjacent right maxillary sinus, and lateral wall of right orbit. Patient underwent magnetic resonance imaging of the spine and computed tomography (CT) of the chest for possible metastasis which showed lesion in lumbar vertebrae and left lung. Patient was planned for CT-guided lung biopsy which proved inconclusive. The patient was further planned for craniotomy and underwent craniotomy with microscopic excision of the mass. Histoimmunochemistry was suggestive of extraskeletal mesenchymal chondrosarcoma. Mesenchymal chondrosarcoma of the brain is highly aggressive tumors which are difficult to differentiate radiologically. Radical excision followed by chemoradiotherapy is optimal treatment of choice.
Keywords
intracranial tumor
extraskeletal mesenchymal chondrosarcoma
metastasis

Introduction
Primary sarcomas involving brain are very uncommon and were first described in the literature by Lichtenstein and Bernstein in 1959.1 Mesenchymal chondrosarcomas were found to the primary tumor of extraosseous sites such as central nervous system but usually originates from soft tissues and bone. Around 2% of chondrosarcomas involving skull base are found in extraskeletal regions.2 The authors described clinical, radiological, and immunohistological features of intracranial extra-axial mesenchymal chondrosarcoma.
Case Report
A 26-year-old man was admitted with features of headache, intermittent episodes of vomiting, decreased vision in right eye for last 15 days. Bilateral cranial nerve examination was normal except right optic nerve with visual acuity of 6/12 on Snellen chart with fundus normal. Motor examination involving muscle strength, reflexes, sensory system, and gait were within normal limits.
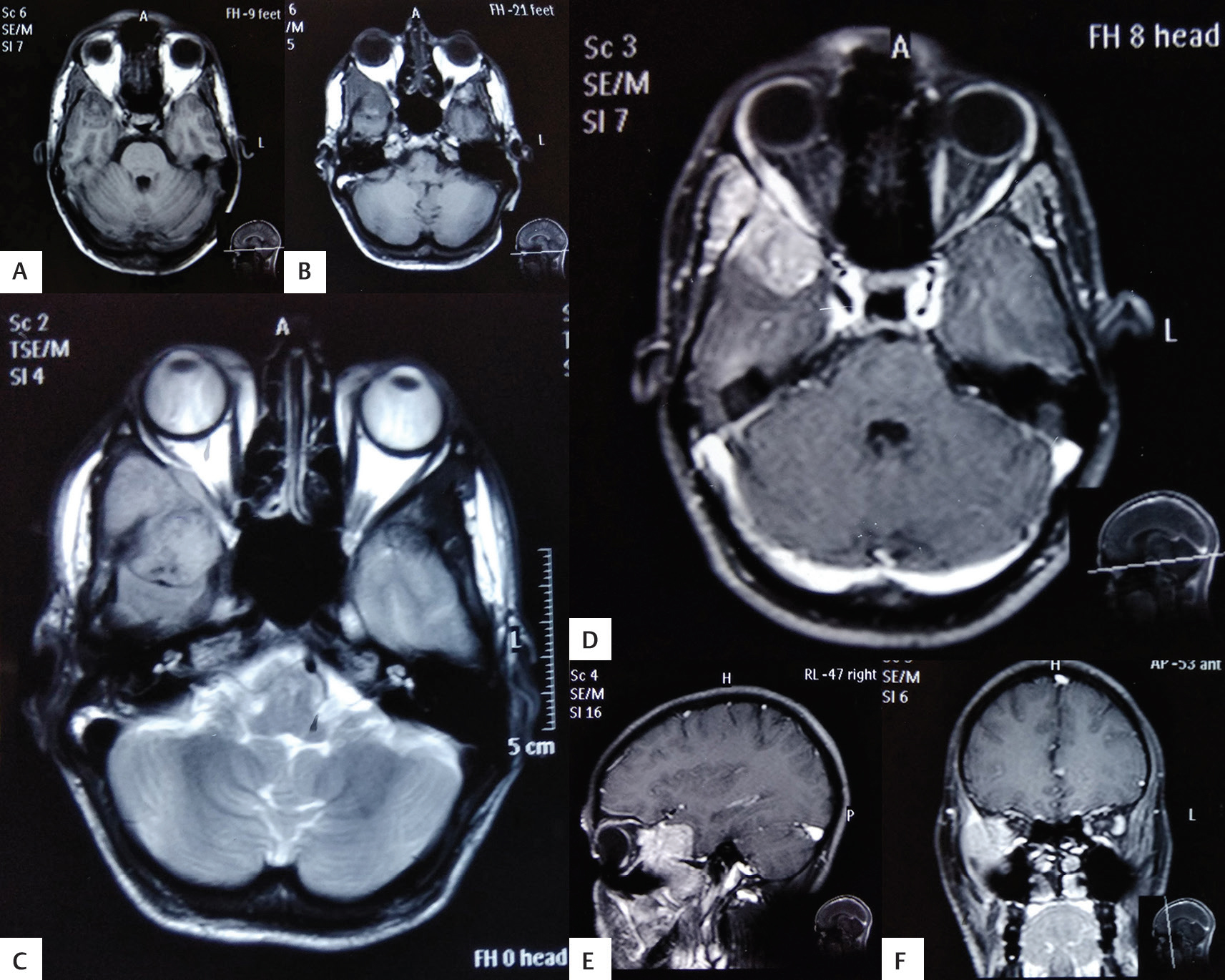
On radiology, computed tomography (CT) of the brain revealed an isodense mass lesion in the right temporal region extending to infratemporal fossa with compression and erosion of right orbital wall. Magnetic resonance imaging (MRI) of the brain revealed a mass in right temporal region involving floor of right middle cranial fossa with patchy enhancement with minimal medial deviation of right lateral wall of right orbit with mild mass effect on right lateral rectus muscle (Fig. 1).
-
Fig. 1 (A-F) Magnetic resonance imaging of the brain revealed mass in right temporal region involving floor of right middle cranial fossa with patchy enhancement with mass effect on right lateral rectus muscle.
Fig. 1 (A-F) Magnetic resonance imaging of the brain revealed mass in right temporal region involving floor of right middle cranial fossa with patchy enhancement with mass effect on right lateral rectus muscle.
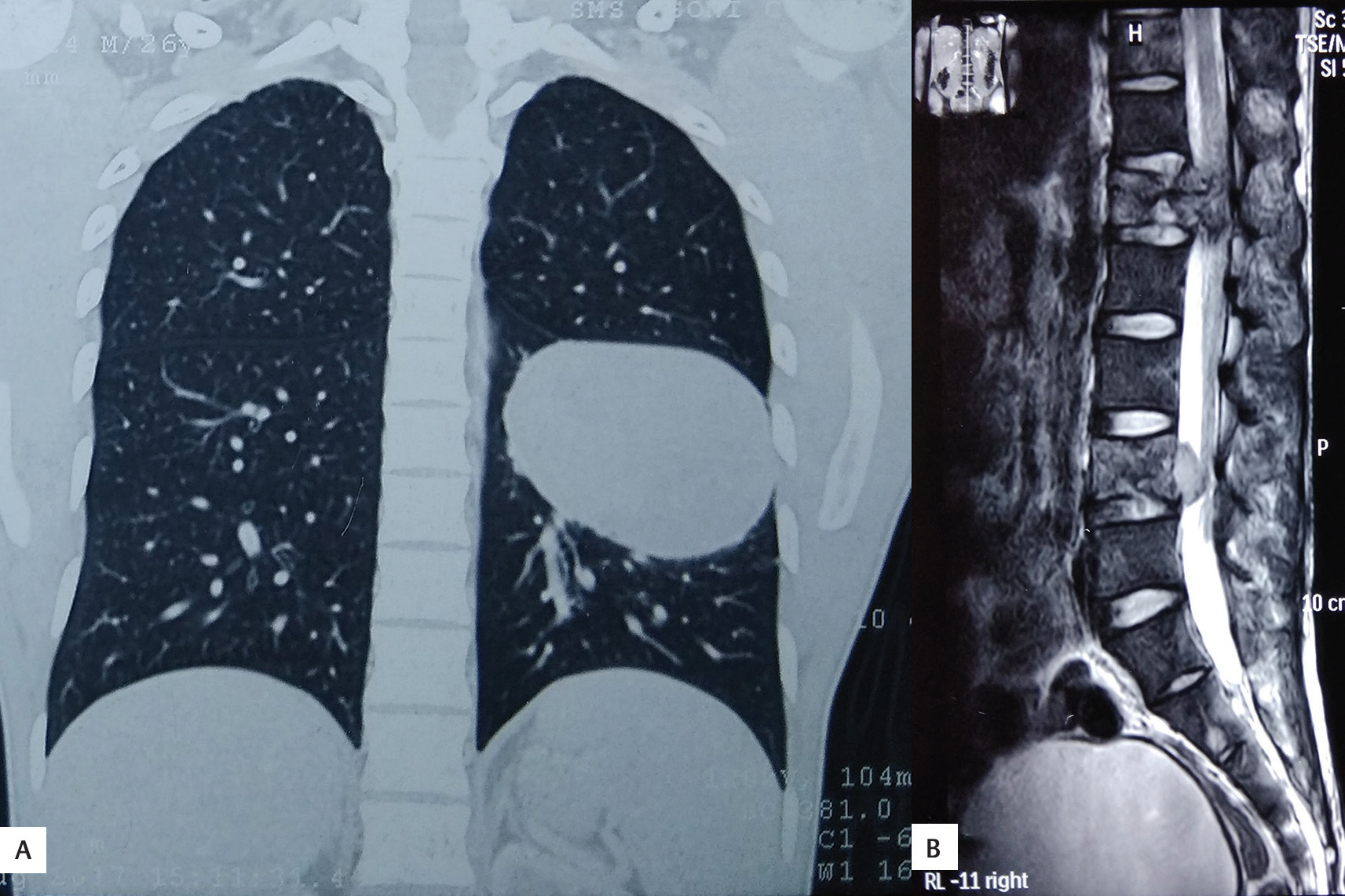
A differential diagnosis of atypical meningiomas, gliomas, or hemangiopericytoma was made preoperatively. The patient underwent CT of the chest and abdomen, and MRI of the spine for possible metastatic lesion which showed a mass in middle and lower zones of left lung and L1 and L4 lumbar vertebra suggestive of partial collapse, possibly metastatic lesions (Fig. 2).
-
Fig. 2 (A, B) Computed tomography of the chest revealed a large lobulated mass in middle and lower zones of left lung and in L1 and L4 lumbar vertebral bodies suggestive of partial collapse.
Fig. 2 (A, B) Computed tomography of the chest revealed a large lobulated mass in middle and lower zones of left lung and in L1 and L4 lumbar vertebral bodies suggestive of partial collapse.
Patient was planned for CT-guided left lung biopsy. Biopsy revealed areas of necrosis with sheets of relatively monomorphic cells with moderate to the scant pale eosinophilic cytoplasm. The nuclei were rounded with mildly irregular nuclear boundaries with chromatin which was coarse at some places with mitotic figures. Immunohistochemistry (IHC) was negative for pan cytokeratin, CD3, and CD20. Overall, biopsy and IHC were nonspecific. So, patient was planned for craniotomy and microscopic excision of mass was done. The tumor was grayish white, firm, partially suckable, highly vascular extradural mass in right temporal region extending to right maxillary sinus, lateral wall of orbit, and attached to dura mater.
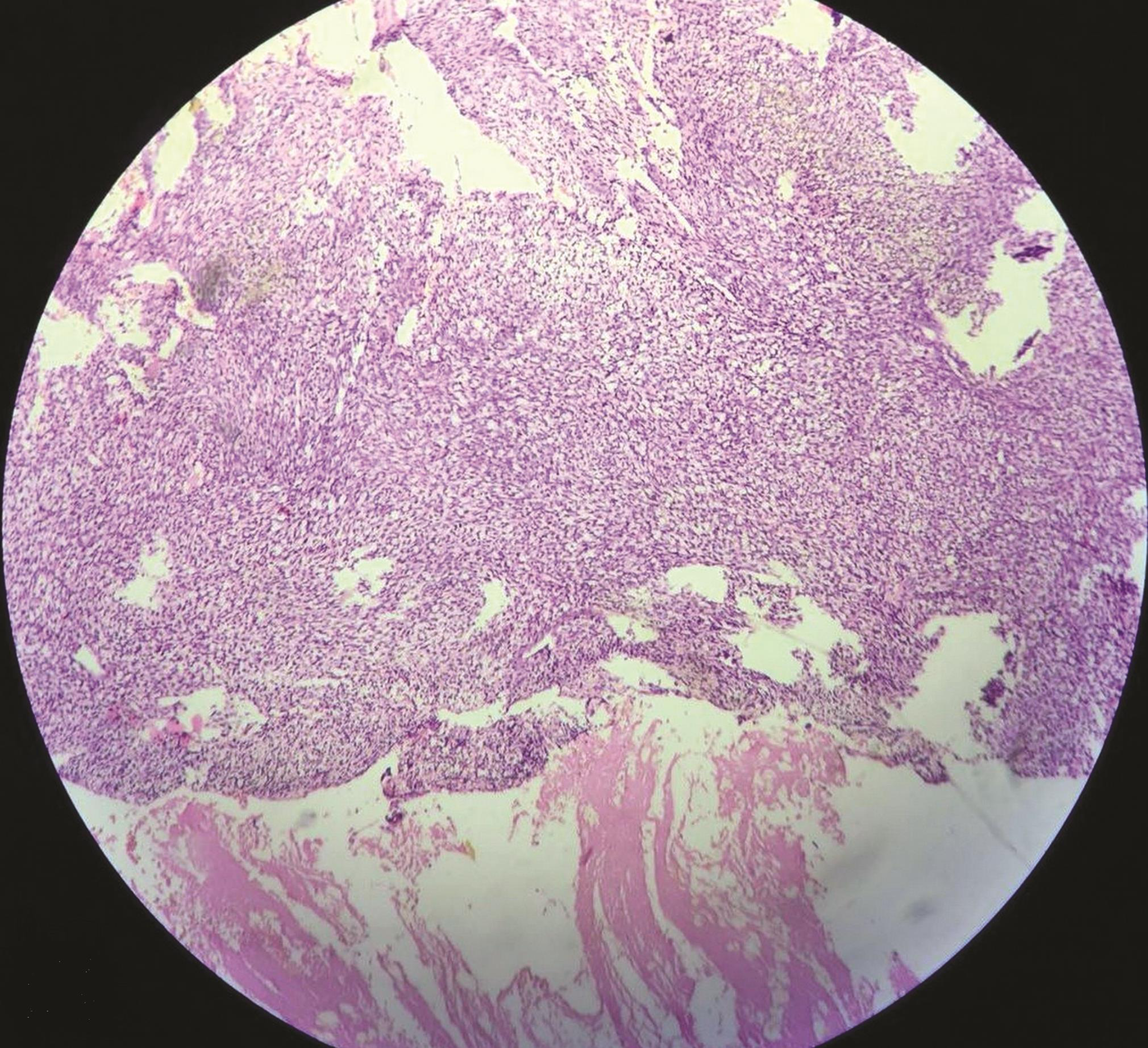
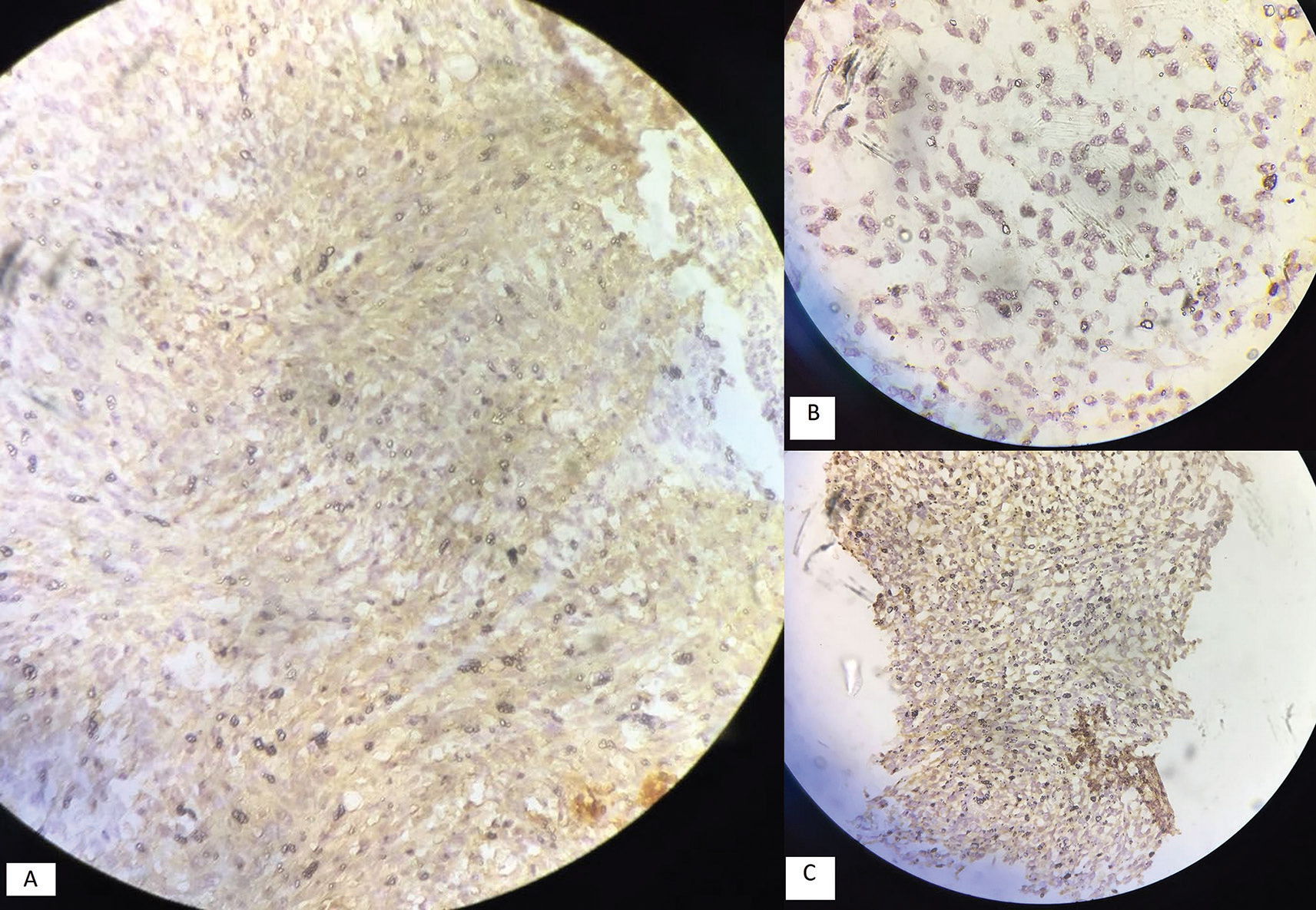
Histopathological examination revealed a spindle to oval cell neoplasm arranged in sheaths with scanty cytoplasm with oval to round nuclei with necrosis. The mitotic count was more than 5/10 high power field with possible diagnosis of mesenchymal tumor, solitary fibrous tumor, hemangiopericytoma grade 3, or Ewing’s sarcoma (Fig. 3). Immunomarkers S100, p53, Bcl2, vimentin, and Ki67/MIB were positive in tumor cells and CK7, CK19, FLE-1, beta-catenin, HMB-45, CK8/18, CD99, epithelial membrane antigen, CD68, CD31, and smooth muscle actin were negative in tumor cells. Immunohistopathology was suggestive of malignant mesenchymal chondrosarcoma (Fig. 4).
-
Fig. 3 Histopathology of tumor tissue (brain) reveals presence of spindle to oval cell neoplasm with presence of necrosis and increased mitotic count.
Fig. 3 Histopathology of tumor tissue (brain) reveals presence of spindle to oval cell neoplasm with presence of necrosis and increased mitotic count.
-
Fig. 4 Immunohistochemistry of tumor tissue (brain) with positive markers of (A) BCL2. (B) P53. (C) S100.
Fig. 4 Immunohistochemistry of tumor tissue (brain) with positive markers of (A) BCL2. (B) P53. (C) S100.
Postoperative CT revealed postoperative changes with no evidence of any residual mass. The patient was advised chemoradiotherapy postoperatively, but the patient did not comply and was lost to follow-up. Patient turned up after 2 months, and was in poor neurological condition and died within few days.
Discussion
Chondrosarcoma of the brain forms only 0.15% of all primary intracranial tumors.3 A mesenchymal chondrosarcoma of the brain with metastasis is the extremely rare variety. Previously, it was thought that these tumors arise only in bones, but 50% of these tumors have origin other than bone such as extraskeletal sites.4 Extraskeletal sites can be soft tissues, muscle, brain, and spinal cord.
Intracranial mesenchymal chondrosarcoma most commonly involve meninges of brain and spinal cord. The first case was reported in 1962 by Dahlin and Hederson.5 These lesions often present as the solitary tumor that grows slowly and present with features of raised intracranial pressure. Intracranial chondrosarcomas are divided into classic and myxoid chondrosarcomas.
Classic chondrosarcomas are mostly seen in males from fourth to sixth decades. It usually involves skull base with no tumor blush on angiography, avascular in nature, locally invasive, and recurrent. Histopathologically, these tumors are characterized by mature cartilage cells without much cellular stroma and more pleomorphism which helps in differentiation from mesenchymal variety.6 In contrast, mesenchymal chondrosarcomas mostly occur in supratentorial region attached to meninges, from second to third decades with no sex preponderence.4 Similarly, our patient presented to us at the age of 26 years.
Clinically, such tumors present as raised intracranial pressure or due to compression on various neighboring anatomical structures. So, the usual presentation of the tumor depends on location and size of the tumor. A longstanding progressive history of a headache, increased intracranial pressure, third nerve palsy, and decreased vision is observed in most patients. In this case, the patient presented to us with features of raised intracranial pressure such as a headache, vomiting, and loss of vision in right eye. These tumors resemble meningioma or hemangiopericytoma and are difficult to differentiate radiologically from meningiomas.
The most origin of chondrosarcoma is from parenchyma, meninges, and base of the skull. The origin of tumor in our case was most probably from the meninges. These tumors are thought to originate from embryonic cartilage cells, fibroblasts, and mesenchymal meningeal cells; however, it is still under investigation.7 However, later theory is more popular. The results of some studies show that even mesenchymal chondrosarcoma is the neoplasm of differentiating premesenchymal chondroprogenitor cells.8
On histology, these tumors are grayish brown, circumscribed, invasive, multilobulated, with rubbery consistency with focal calcification and high vascularity. These tumors have two differentiating appearances of undifferentiated high cellular mesenchymal cells, and benign appearing cartilage cells with abrupt transition between the two.
On IHC, these cells are highly sensitive for vimentin, S100 protein, and negative for glial fibrillary acidic protein and neuron-specific enolase. Similarly, in our case, overall morphology favored a malignant mesenchymal neoplasm as discussed previously.
Mesenchymal chondrosarcoma of the brain can metastasize to lumbar vertebrae, ribs, heart, and several intra-abdominal structures.9 Similarly, our patient presented to us with metastasis in lumbar vertebrae and left lung. The 5- and 10-year survival rates observed in various case series have between 42 to 68 and 28 to 32%,10 respectively. Our patient was advised chemoradiotherapy, but lost to follow-up and expired after 2 months.
Radical surgical excision can be combined with neoadjuvant chemotherapy for these tumors. Tumors with inadequate surgical excision and unresectable tumors may benefit from radiation with or without chemotherapy. Adjuvant therapy is still offered to all patients because of the rarity of the tumor, propensity for recurrences, metastasis, and death.
Conclusion
Mesenchymal chondrosarcoma are highly aggressive neoplasm in young adults and are difficult to differentiate on the basis of radiology and histopathology. Histopathological examination, IHC, and expert pathologist are key to diagnosis of such lesions. Radical excision is optimal treatment of choice. Postoperatively, radiation with or without chemotherapy can be given to patients to reduce recurrence. Patients with inadequate resection margins after surgery and unresectable tumors can be offered chemoradiotherapy. As these tumors are highly aggressive and invasive with potential for recurrences and metastasis, the patient should be kept under regular follow-up.
Conflict of Interest
None declared.
References
- Unusual benign and malignant chondroid tumors of bone. A survey of some mesenchymal cartilage tumors and malignant chondroblastic tumors, including a few multicentric ones, as well as many atypical benign chondroblastomas and chondromyxoid fibromas. Cancer. 1959;12:1142-1157.
- [Google Scholar]
- Mesenchymal chondrosarcoma: clinicopathologic study of 20 cases. Arch Pathol Lab Med. 2012;136(1):61-75.
- [Google Scholar]
- Primary cranial and intracranial chondrosarcoma. A survey. Acta Neurochir (Wien). 1985;78:123-132. (3-4)
- [Google Scholar]
- Meningeal mesenchymal chondrosarcoma: report of 8 cases with review of the literature. Cancer. 1978;42(6):2744-2752.
- [Google Scholar]
- Mesenchymal chondrosarcoma. Further observations on a new entity. Cancer. 1962;15:410-417.
- [Google Scholar]
- Intracranial extraskeletal mesenchymal chondrosarcoma: case report. Neurosurgery. 2000;46(1):207-211, discussion 211–212.
- [Google Scholar]
- Central nervous system mesenchymal chondrosarcoma. Case report. J Neurosurg. 1984;61(2):375-381.
- [Google Scholar]
- Cell differentiation and matrix gene expression in mesenchymal chondrosarcomas. Am J Pathol. 2000;156(4):1327-1335.
- [Google Scholar]
- Intracranial mesenchymal chondrosarcoma: a case report and literature review. Childs Nerv Syst. 1993;9(5):295-299.
- [Google Scholar]
- Primary meningeal mesenchymal chondrosarcoma. Arch Pathol Lab Med. 1979;103(5):239-243.
- [Google Scholar]




