
Translate this page into:
Joubert Syndrome with Orofacial Digital Features
Address for correspondence: Dr. Minoo Sharma, Department of Pediatrics and Physiology, Indira Gandhi Medical College, Shimla - 171 001, Himachal Pradesh, India. E-mail: minoo01sharma@gmail.com
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Joubert syndrome (JS) is an autosomal recessive inherited disorder characterized by hypotonia, cerebellar vermis hypoplasia, ocular abnormalities (e.g., pigmentary retinopathy, oculomotor apraxia, and nystagmus), renal cysts, and hepatic fibrosis. Respiratory abnormalities, as apnea and hyperpnea, may be present, as well as mental retardation. Since the clinical findings of JS are quite heterogeneous, determination of radiological findings is essential.
Keywords
Cerebellar malformation
ciliopathy
Joubert syndrome
magnetic resonance imaging
molar tooth sign

INTRODUCTION
Joubert syndrome (JS) was first described by Marie Joubert in 1969 in four French-Canadian siblings with hypotonia, ataxia, abnormal eye movements, and alternating apnea/hyperpnea.[1] It would take another three decades for the molar tooth sign (MTS) correlation on magnetic resonance imaging (MRI) to emerge in this now eponymously named JS.[2]
The prevalence of JS and related disorders (JSRDs) has not been determined. Many authors use a range between 1:80,000 and 1:100,000, but this may represent an underestimate.[34] There is a relatively high prevalence of JSRD in the French-Canadian population.[5]
We report a case of JS with orofacial digital features.
CASE REPORT
A 3-year-old male presented to us with a history of developmental delay and extra digits. The child was the product of nonconsanguineous marriage. Antenatal history was uneventful and there was no history suggestive of birth asphyxia. There was no history of abnormal respiratory pattern, apnea, or abnormal movements.
On examination, the child was conscious and alert, and vital signs were stable. There were no neurocutaneous markers and no evidence of facial dysmorphism. On developmental examination, the child was found to be delayed in all the four domains of gross motor, fine motor, speech, and person and social. His developmental age was about 9–12 months. There was no evidence of cranial nerve palsies, and on motor system examination, the child was grossly hypotonic and deep tendon reflexes were normal. There were soft-tissue masses on the dorsum of the tongue [Figure 1a] and also had polydactyly with six digits in bilateral hands and left foot [Figure 1b]. The child also had protein energy malnutrition Grade II and signs of rickets; rest of the clinical examinations were normal. On investigation, complete blood count, thyroid function test, arterial blood gas, renal function test electrolytes, and liver function test were normal. In view of development delay, MRI was done and it revealed vermis hypoplasia with deep interpeduncular fossa with thick and elongated superior cerebellar peduncle and midbrain giving “molar tooth” appearance [Figure 2a]. In addition, there was heterotrophic gray matter in the right centrum semiovale and periventricular white matter [Figure 2c].
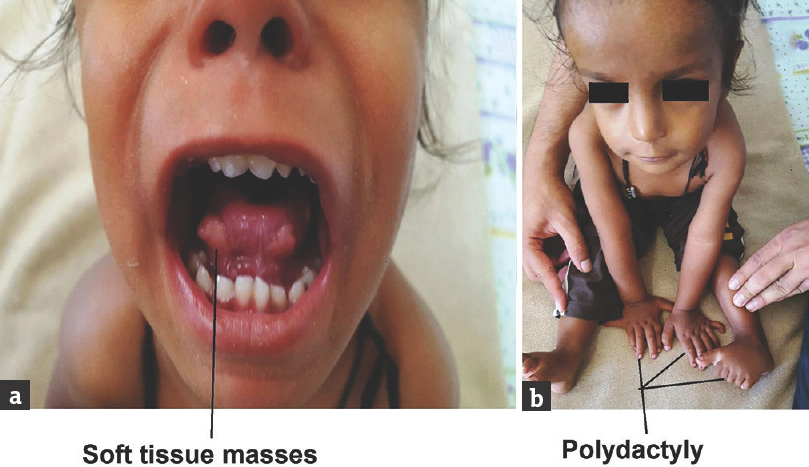
- Index case with tongue soft-tissue masses (a) and polydactyly (b)
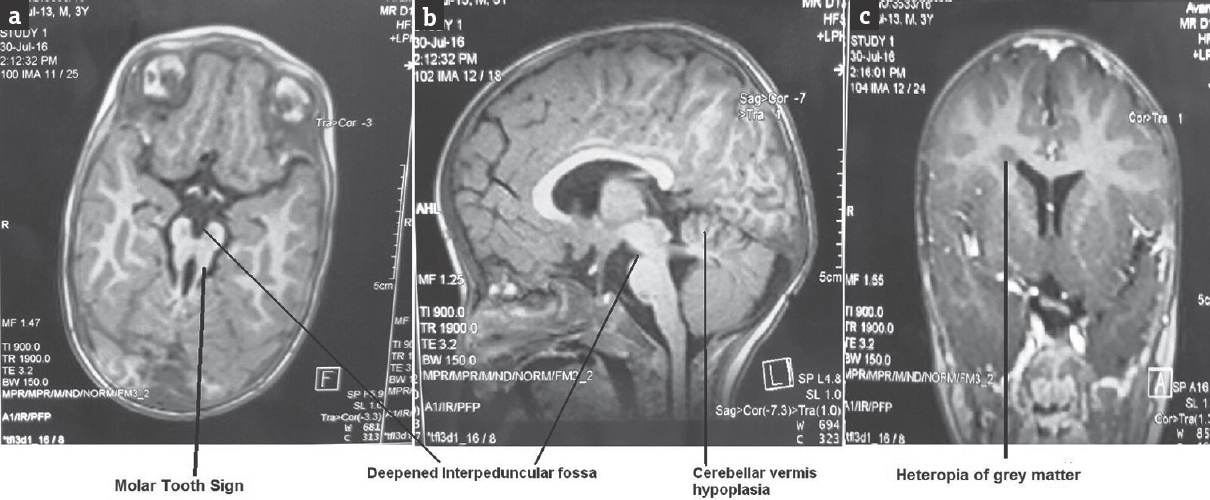
- Magnetic resonance imaging brain showing molar tooth sign (a), deepened interpeduncular fossa, cerebellar vermis hypoplasia (b), and heterotopias of gray matter (c)
Hence, in view of the development delay, polydactyly, soft-tissue masses on the tongue, and the presence of MTS on MRI, possibility of Joubert Syndrome with orofacial digital (JS-OFD) features was kept.
DISCUSSION
Classic JS is diagnosed by three primary findings of a distinctive cerebellar and brainstem malformation called the MTS, hypotonia, and developmental delays. Often, these findings are accompanied by episodic tachypnea or apnea[6] and/or atypical eye movements.[7] In general, the breathing abnormalities improve with age, truncal ataxia develops over time, and acquisition of gross motor milestones is delayed.[8] The designation JSRDs is used to describe individuals with JS who have additional findings.
JS with Orofacial digital (JS-OFD) features. Polydactyly is described in 8%–19% of probands.[14] Polydactyly can be unilateral or bilateral and is often postaxial as seen in our patient. In addition to polydactyly, individuals with OFD often have cerebellar vermis hypoplasia, oral frenula, tongue lobulations, or hamartomas as seen in the renal and cardiac involvement.[9]
JS with retinal disease (JS-Ret) is characterized by a pigmentary retinopathy. It can occasionally be severe with neonatal onset of congenital blindness. Ocular colobomas are most often chorioretinal and may be associated with hepatic fibrosis.[10]
JS with renal disease (JS-Ren) has been described as a continuum of disease varying from chronic tubulointerstitial nephropathy in the form of juvenile nephronophthisis to nephrocystic disease which is clinically and morphologically similar to autosomal recessive polycystic kidney disease.[9]
JS with oculorenal disease (JS-OR) has been described with features of both renal and retinal impairment.[4]
JS with hepatic disease (JS-H) is characterized by hepatic fibrosis which is usually progressive but rarely symptomatic at birth. Hepatic fibrosis was observed in 18% of individuals with JSRD in one cohort.[1]
JSRDs follow autosomal recessive inheritance and are genetically heterogeneous with one locus pointing to chromosome 9q. Ten causative genes have been recognized so far, with every single one encoding for proteins of the primary cilium or the centrosome, making JSRD a part of an expanding group of diseases called “ciliopathies.”[4611]
A high-quality MRI scan, including axial, coronal, and sagittal views (ideally with 3-mm axial planes through the midbrain and pons) is required for establishing the diagnosis. MTS is the classical sign that establishes the diagnosis of JS. This molar tooth appearance on MRI brain is due to cerebellar vermis hypoplasia, deepened interpeduncular fossa [Figure 2b], and elongated superior cerebellar peduncles. In addition to the MTS, images should be reviewed for other findings, such as polymicrogyria, callosal abnormalities, heterotopias, and evidence of ventriculomegaly or a posterior fossa fluid collection suggestive of Dandy–Walker malformation.
An electroencephalogram (EEG) is indicated if clinical seizures are suspected. Polysomnogram in all children diagnosed under the age of 12 months to differentiate breathing pauses due to obstructive events (such as a hypotonic airway or enlarged tonsils) from centrally mediated brainstem dysregulation more typical of JS. As the majority of infants will not manifest renal cystic disease or liver fibrosis at this young age, interval re-evaluation with abdominal ultrasonography is advised.
Treatment is usually supportive and includes a multidisciplinary approach to management. Infants and children with abnormal breathing patterns should be considered for apnea monitoring if the abnormality is severe. Supportive therapy may include stimulatory medications such as caffeine or supplementary oxygen, particularly in the newborn period. In rare cases, mechanical support and/or tracheostomy may be considered in a child with severe respiratory dysfunction. Nasogastric feeding tubes or gastrostomy tube placement was used for feeding in children with severe dysphagia. Seizures should be evaluated by EEG and treated with standard antiepileptic drugs under the management of a neurologist. Corrective surgery can be done for polydactyly. Tongue tumors that impair normal swallowing or cause respiratory obstruction may require surgical resection.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- Joubert syndrome: Insights into brain development, cilium biology, and complex disease. Semin Pediatr Neurol. 2009;16:143-54.
- [Google Scholar]
- “Joubert syndrome” revisited: Key ocular motor signs with magnetic resonance imaging correlation. J Child Neurol. 1997;12:423-30.
- [Google Scholar]
- The birth prevalence of Joubert syndrome: A population based study in the Netherlands. Eur J Hum Genet. 2007;15:68.
- [Google Scholar]
- Clinical features and revised diagnostic criteria in Joubert syndrome. J Child Neurol. 1999;14:583-90.
- [Google Scholar]
- Joubert Syndrome and Related Disorders. 2003. GeneReviews®. Seattle (WA): University of Washington, Seattle; 1993-2016; Available from: https://www. ncbi.nlm.nih.gov/books/NBK1325/#
- [Google Scholar]
- Oral-facial-digital syndrome type VI (Váradi syndrome): Further clinical delineation. Am J Med Genet. 1990;35:360-9.
- [Google Scholar]
- Clinical and molecular features of Joubert syndrome and related disorders. Am J Med Genet C Semin Med Genet. 2009;151C:326-40.
- [Google Scholar]
- Clinical nosologic and genetic aspects of Joubert and related syndromes. J Child Neurol. 1999;14:660-6.
- [Google Scholar]




