
Translate this page into:
Epileptic Spasms in an Infant with Incontinentia Pigmenti: Report of a Rare Case with Brief Review of the Literature
Prateek Kumar Panda, DM Department of Pediatrics, All India Institute of Medical Sciences Virbhadra Road, Rishikesh, Uttarakhand 249203 India drprateekpanda@gmail.com
This article was originally published by Thieme Medical and Scientific Publishers Private Ltd. and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Abstract
Incontinentia pigmenti (IP) or Bloch–Sulzberger’s disease is a rare neurocutaneous syndrome with dermatological, neurologic, and systemic manifestations including retinal, dental and hair abnormalities. It follows X-linked dominant inheritance and predominantly affects female children. The characteristic evolution of skin lesions in four stages is a hallmark diagnostic feature of the disease. The pigmented lesions of IP are usually distributed in linear streaks, macular whorls, reticulated patches, and flecks along the Blaschko lines. Neurologic morbidities are found in a considerable proportion of affected children, and the spectrum includes seizures, neuromotor impairment, microcephaly, developmental delay, and intellectual disability. Seizures are reported in 10% to 25% of children with IP in various previous clinical studies. The majority of these children had seizures in the neonatal period or early infancy, and focal–clonic seizure is the commonest observed semiology. However, there are only a few case reports of infants with IP with epileptic spasms. In this report, the clinical course of a 6-month-old girl with IP and epileptic spasms has been described, who responded favorably to treatment with adrenocorticotropic hormone injection. Clinicians managing children with IP should be aware of their predisposition to develop epileptic spasms and consider neuroimaging, electroencephalogram, and other investigations accordingly.
Keywords
neurocutaneous syndrome
incontinentia pigmenti
Blaschko lines
West’s syndrome
modified hypsarrhythmia

Introduction
Incontinentia pigmenti (IP) or Bloch–Sulzberger’s disease is a rare neurocutaneous syndrome with dermatological, neurologic, and systemic manifestations including retinal, dental, and hair abnormalities. It follows X-linked dominant inheritance and predominantly affects female children. As such, it is lethal in most affected male fetuses and predominantly affects female children.1 It was first reported in 1906,2 and the classic characteristics of this disorder include pigmentary skin changes along with other organ system abnormalities. Systemic abnormalities predominantly involve the brain, eyes, and teeth.3 Other rare manifestations such as breast anomalies (breast hypoplasia and supernumerary nipples) may occur in 1% of patients. Similarly, approximately 14% of children have skeletal and structural anomalies such as somatic asymmetry, hemivertebrae, scoliosis, spina bifida, syndactyly, ear anomalies, extra ribs, skull deformities, primary pulmonary hypertension, and cardiopulmonary failure. It is an extremely rare disease, with an estimated prevalence being around 0.2 per 100,000 births.2 It is because of mutations involving a gene located on chromosome Xq28, which codes for the inhibitor of nuclear factor κB kinase γ (IKBKG), previously named NEMO.4
The characteristic evolution of skin lesions in four stages is a hallmark diagnostic feature of the disease. The pigmented lesions of IP are usually distributed in linear streaks, macular whorls, reticulated patches, and flecks, along the Blaschko lines.4 However, not all stages may be observed in every single case.4 Eye abnormalities mostly involve the retina. The retinal abnormalities are predominantly due to vascular occlusion and usually present with neovascularization, hemorrhages, exudative and tractional detachments, and rarely the absence of the foveal pit. Abnormalities of teeth and hair, as well as central nervous system (CNS) anomalies leading to seizure, neuromotor impairment, developmental delay, and intellectual disability, are also observed in a considerable proportion.4 Although West’s syndrome is relatively more prevalent in other neurocutaneous syndromes such as tuberous sclerosis and neurofibromatosis, only a few case reports describe epileptic spasms in children with IP.5 In this report, we describe an infant with IP and epileptic spasms and successfully treated with adrenocorticotropic hormone (ACTH) injection.
Case Summary
Clinical Presentation
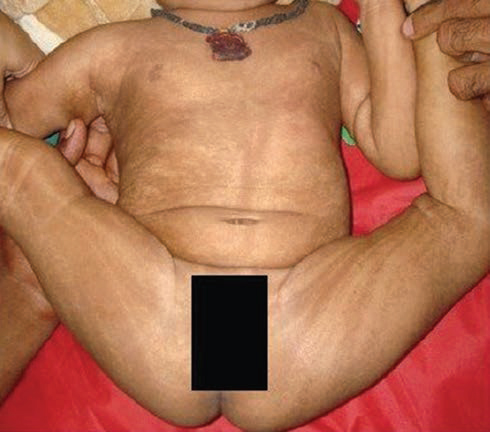
A 6-month-old girl presented with multiple paroxysmal events in the form of sudden onset flexion of both upper and lower extremities followed by tightening for few seconds (1–2 seconds), occurring predominantly at sleep–wake transition. These events were confirmed to be epileptic spasms after reviewing the video records. At the time of presentation, the frequency of epileptic spasm was 10 to 15 spasms per cluster and approximately 6 to 10 clusters per day. Only occasional single epileptic spasms used to occur in the awake state. The child also had developmental delay, microcephaly (Z-score ≤ 3), and linear and whorled hyperpigmented lesions over the trunk and extremities (Fig. 1) on general physical examination. The child had achieved partial head control, was able to recognize the mother and, had a social smile. However, she did not achieve rolling over or bidextrous grasp to hold objects, thus leading to a developmental age of about 3 months. On leading questioning, the caregivers revealed the perinatal period of the child was uneventful otherwise. However, from the first week onward, erythematous lesions appeared over trunk and extremities. Many of these lesions get converted to vesicles and blisters. At around 4 months of age, these lesions were replaced by verrucous, hyperkeratotic lesions, and for the last few weeks, hyperpigmented streaks appeared in place of these verrucous lesions.
-
Fig. 1 Clinical photograph of the child showing whorled hyperpigmented lesions on the trunk and extremities along the Blaschko lines.
Fig. 1 Clinical photograph of the child showing whorled hyperpigmented lesions on the trunk and extremities along the Blaschko lines.
Diagnostic Evaluation and Management
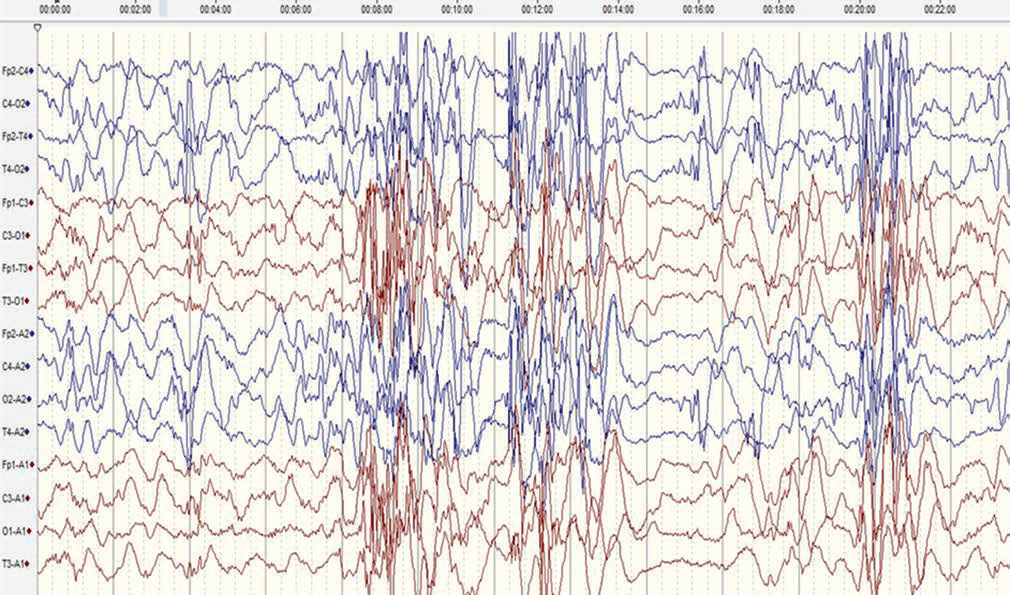
The electroencephalogram (EEG) of the child revealed modified hypsarrhythmia (Fig. 2), and the diagnosis of West’s syndrome was confirmed. Magnetic resonance imaging (MRI) of the brain revealed mild cerebral atrophy. The ophthalmic evaluation suggested retinal detachment in the left eye only. Complete blood count revealed eosinophilia (11% of total leucocyte count). There was no family history suggestive of any such symptoms. The epileptic spasms subsided after 10 days with the institution of ACTH injection used in an escalating dose up to 75 IU/m2. Repeat EEG after 4 weeks showed resolution of hypsarrhythmia and only occasional multifocal discharges. In view of the constellation of clinical findings compatible with IP, next-generation sequencing was performed, which revealed a missense pathogenic mutation in the NEMO gene on X chromosome. Thus, the diagnosis of IP with West’s syndrome was concluded.
-
Fig. 2 A sedated electroencephalogram (international 10–20 system, bipolar montage, sensitivity of 7 µV/mm and sweep speed of 30 mm/second, low-frequency filter of 1 Hz, and high-frequency filter of 35 Hz) (before adrenocorticotropic hormone therapy) showing paucity of sleep markers and generalized bursts of high-amplitude spikes, polyspikes, and slow-wave activity alternating with periods of voltage attenuation, suggestive of modified hypsarrhythmia. Frequent polyspikes and waves were also noted from the right posterior head region.
Fig. 2 A sedated electroencephalogram (international 10–20 system, bipolar montage, sensitivity of 7 µV/mm and sweep speed of 30 mm/second, low-frequency filter of 1 Hz, and high-frequency filter of 35 Hz) (before adrenocorticotropic hormone therapy) showing paucity of sleep markers and generalized bursts of high-amplitude spikes, polyspikes, and slow-wave activity alternating with periods of voltage attenuation, suggestive of modified hypsarrhythmia. Frequent polyspikes and waves were also noted from the right posterior head region.
Follow-Up
After epileptic spasms subsided, the child was operated for retinal detachment in the left eye. The child did not have any other nail, hair, or skeletal anomalies. The child was continued on early stimulation therapy for developmental delay. The parents were also provided with appropriate genetic counseling. On follow-up at 6 months, the child was seizure-free, off antiepileptic drugs, and gaining milestones.
Discussion
Our case describes new-onset epileptic spasms in a 6-month-old girl with IP and without previous seizures, who responded favorably to ACTH injection. Existing literature describes only four cases of West’s syndrome in children with IP. All of the cases have evidence of vascular insult to the brain in neuroimaging, unlike our case, in which only mild cerebral atrophy was found. Only in one case described by Simonsson in 1972, no neuroimaging was performed. But in this case’s follow-up EEG at 1 ½ year showed slowing over right hemisphere electrodes, indicating probably underlying structural abnormality. The other contrasting finding was that most of these children had previously neonatal-onset unprovoked seizures of different semiology that later on evolved to West’s syndrome. Only in the case described by Simonsson, like the index case, there was de novo onset epileptic spasm at 6½ months, and the case responded favorably to ACTH injection with complete resolution in spasms.5 In our case, de novo onset of epileptic spasms, with only mild cerebral atrophy in brain MRI, indicates there is some genetic predisposition over and above the structural abnormalities of the brain, which predisposes toward epileptogenesis. Clinicians treating children with IP need to be aware of the possibility of epileptic spasms in these children, although it is extremely rare.
The exact pathophysiology of the CNS anomalies leading to the seizure and developmental delay is yet to be elucidated, but, usually, it is attributed to vascular injury, inflammatory mechanisms, or disturbed apoptosis during development.6 Hadj-Rabia et al in a large series of 40 children with IP demonstrated neurologic features in 32% of children. Out of the 10 children with seizures, most had seizure onset in the neonatal period. Two children with severe and extensive dermatological manifestations had generalized seizures and vascular damage with thalamic hemorrhage, leading to death. Neuroimaging was abnormal in approximately 70% of children with neurologic symptoms.6 Predominant neuroimaging abnormalities reported in this study ranged from cerebral atrophy only to focal or extensive hemorrhagic necrosis. Porencephaly, hypoplasia of the corpus callosum, isolated enlargement of lateral ventricles, basal ganglia hemorrhages, and white matter signal changes were other neuroimaging findings noticed in the study.6
Carney had also documented similar findings of CNS abnormalities in approximately 30.5% out of 653 children with IP.7 However, subsequent authors concluded that some of these children had hypomelanosis of Ito rather than IP.8 Abe et al reported “pseudoencephalitis”-like initial neurologic manifestation with coma and apnea in two infants due to acute infarctions caused by ischemic cerebrovascular accidents.8
In consonance with the aforementioned findings, an analysis of 18 separate studies by Minić et al also revealed seizure as the commonest neurologic symptom (present in 13–25% of children).9 In this series also, seizures occurred in the neonatal period or in early infancy in the majority of cases. However, seizure onset occurred in some children up to 10 years of age. The most predominant seizure semiology observed was focal–clonic type. Recurrence of seizures occurred in around 40% cases. In most of the cases, EEG was abnormal, but no pattern specific for IP could be ascertained.9 Neuroimaging abnormalities were more common in patients with seizures as compared with those without seizures.9 However, the patterns of abnormalities in both groups were similar and like previous studies were suggestive of underlying vascular insufficiency. Periventricular leukomalacia often with gliotic changes and cavitation, basal ganglia damage, and diffuse hemorrhagic necrosis were observed in children with IP in various studies.10
As mentioned earlier, there are reports of only four different children with IP and West’s syndrome in the existing literature.5 11 All three children described by Poziomczyk et al had early-onset seizures from the first week of life, unlike the index case described earlier.11 All three cases had abnormal neuroimaging, hypsarrhythmia in EEG, and responded to corticosteroids or ACTH injection, like the index case. Other electroclinical epileptic syndromes have not been described until now in children with IP.
Conclusion
Clinicians managing children with IP should be aware of their predisposition to develop seizures and consider neuroimaging, EEG, and other investigations accordingly. Although West’s syndrome is quite rare in these children, still early recognition and institution of proper treatment such as corticosteroid or ACTH injection are imperative for favorable long-term neurodevelopmental outcome in these children.
Conflict of Interest
None declared.
Funding None.
References
- X inactivation as a mechanism of selection against lethal alleles: further investigation of incontinentia pigmenti and X linked lymphoproliferative disease. J Med Genet. 1992;29(9):608-614.
- [Google Scholar]
- The International Incontinentia Pigmenti (IP) Consortium. Genomic rearrangement in NEMO impairs NF-kappaB activation and is a cause of incontinentia pigmenti. Nature. 2000;405 (6785):466-472.
- [Google Scholar]
- Incontinentia pigmenti: a review and update on the molecular basis of pathophysiology. J Am Acad Dermatol. 2002;47(2):169-187, quiz 188–190.
- [Google Scholar]
- Incontinentia pigmenti, Bloch-Sulzbergers syndrome, associated with infantile spasms. Acta Paediatr Scand. 1972;61(5):612-614.
- [Google Scholar]
- Clinical study of 40 cases of incontinentia pigmenti. Arch Dermatol. 2003;139(9):1163-1170.
- [Google Scholar]
- Incontinentia pigmenti. A world statistical analysis. Arch Dermatol. 1976;112(4):535-542.
- [Google Scholar]
- Early infantile manifestations of incontinentia pigmenti mimicking acute encephalopathy. Brain Dev. 2011;33(1):28-34.
- [Google Scholar]
- Systematic review of central nervous system anomalies in incontinentia pigmenti. Orphanet J Rare Dis. 2013;8:25.
- [Google Scholar]
- Intracranial assessment of incontinentia pigmenti using magnetic resonance imaging, angiography, and spectroscopic imaging. Arch Pediatr Adolesc Med. 1995;149(5):573-580.
- [Google Scholar]




