
Translate this page into:
Chromosome Xp22.3 deletion syndrome with X-linked ichthyosis, Kallmann syndrome, short stature, generalized epilepsy, hearing loss, attention deficit hyperactivity disorder, and intellectual disability – A rare report with review of literature
 , Sujatha Manjunathan1, Veena Laxmi1, Rahul Gupta1, Ashna Kumar1, Arun Sree Parameswaran1, Achanya Palayullakandi2, Anil Budania3, Kuldeep Singh1
, Sujatha Manjunathan1, Veena Laxmi1, Rahul Gupta1, Ashna Kumar1, Arun Sree Parameswaran1, Achanya Palayullakandi2, Anil Budania3, Kuldeep Singh1
*Corresponding author: Lokesh Saini, Department of Pediatrics, All India Institute of Medical Sciences, Jodhpur, Rajasthan, India. drlokeshsaini@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Gunasekaran P, Saini L, Rajial T, Manjunathan S, Laxmi V, Gupta R, et al. Chromosome Xp22.3 deletion syndrome with X-linked ichthyosis, Kallmann syndrome, short stature, generalized epilepsy, hearing loss, attention deficit hyperactivity disorder, and intellectual disability – A rare report with review of literature. J Neurosci Rural Pract. 2024;15:425-30. doi: 10.25259/JNRP_467_2023
Abstract
Chromosome Xp22.3 deletion syndrome is a very rare contiguous gene deletion syndrome with variable phenotype due to the deletion of genes from the distal short arm of the X chromosome (Xp), including the short-stature homeobox (SHOX), anosmin-1 (ANOS1), arylsulfatase (ARSL), neuroligin-4 (NLGN4), and steroid sulfatase (STS) genes. We have reviewed the available literature on the chromosome Xp22.3 deletion syndrome. A 10-year-old boy presented with global developmental delay, generalized epilepsy, decreased hearing, and hyperactivity. He had no significant family history. Examination revealed microcephaly, short stature, and dry and scaly skin lesions on the trunk. He had thick arched eyebrows, a depressed nasal bridge, a long philtrum, high arched palate, retrognathia, brachytelephalangy, brachymetatarsia, and mild scoliosis. Brainstem-evoked response audiometry testing revealed moderate hearing loss. Magnetic resonance imaging showed cerebellar tonsillar ectopia. Clinical exome sequencing revealed a likely pathogenic contiguous deletion (~8.10 Mb) spanning genomic location chrX:g.(_630898)_(8732037_)del encompassing ANOS1, ARSL, NLGN4X, SHOX, and STS genes. We have reviewed the available literature for reported associations of Chromosome Xp22.3 deletion syndrome and report a novel association of X-linked ichthyosis, Kallmann syndrome, global developmental delay, short stature, bilateral hearing loss, generalized epilepsy, attention deficit hyperactivity disorder, and intellectual disability.
Keywords
Chromosome Xp22.3 deletion syndrome
X-linked ichthyosis
Kallmann syndrome
Short stature
Generalized epilepsy

INTRODUCTION
Chromosome Xp22.3 deletion syndrome is a very rare contiguous gene deletion syndrome with variable phenotype, depending on the large terminal or interstitial codeletions of adjacent genes that are physically clustered. This contiguous gene syndrome is due to the deletion of genes from the distal short arm of the X chromosome (Xp), including the short-stature homeobox (SHOX), anosmin-1 (ANOS1), arylsulfatase (ARSL), neuroligin-4 (NLGN4), and steroid sulfatase (STS) genes.[1] This syndrome is characterized by X-linked ichthyosis (XLI), Kallmann syndrome (KS), short stature, generalized epilepsy, neurodevelopmental disorders including autism spectrum disorder (ASD), attention deficit hyperactivity disorder (ADHD), and intellectual disability (ID), sensorineural hearing loss, strabismus, and bony deformities including chondrodysplasia punctata (CDP), obesity, and ocular albinism.[1-3] The other dysmorphism features reported are frontal bossing, hypertelorism, depressed nasal bridge, high-arched palate, retrognathia, and flat occiput.[4,5] Based on the deletion lengths, the clinical phenotypes occur independently from each other or in various combinations as a contiguous gene syndrome.[6] Only about 34 cases have been reported in the literature. Here, we report a case of Xp22.3 deletion syndrome (8.1 Mb contiguous deletion), including ANOS1, ARSL, NLGN4X, SHOX, and STS genes, along with a review of available literature [Table 1].
| S. No | Location | Genes | MIM | Phenotype | Phenotype MIM | |
|---|---|---|---|---|---|---|
| 1. | Xp22.31 | ANOS1 | Anosmin-1 | 300836 | Kallmann syndrome | 308700 |
| 2. | Xp22.31 | STS | Steroid sulfatase | 300747 | Ichthyosis | 308100 |
| 3. | Xp22.32 | NLGN4X | Neuroligin-4 | 300427 | ASD, ID | 300495 |
| 4. | Xp22.33 | ARSL | Arylsulfatase-L | 300180 | Chondrodysplasia punctata, Short stature | 302950 |
| 5. | Xp22.33 | SHOX | Short stature homeobox | 312865 | Short stature LWD, MD | 300582 |
ASD: Autism spectrum disorder; ID: Intellectual disability; LWD: Leri-Weill dyschondrosteosis; MD: Madelung deformity; MIM: Online Mendelian inheritance in man.
CASE PRESENTATION
A 10-year-old boy presented with global developmental delay, decreased hearing since birth, and hyperactivity. He also had a history of generalized tonic-clonic seizures since five years of age, with 1–2 episodes/year. He was first born to third-degree consanguineously married parents. The antenatal and perinatal periods were unremarkable. There was no significant family history. On examination, he had a height of 116 cm (−3.5Z, stunting), a weight of 25.2 Kg (−1.3Z), body mass index of 18.7 kg/m2, and a head circumference of 53 cm. The upper-to-lower segment ratio (US: LS) was 0.93. The mid-parental height was 159 cm. He had coarse facies with thick arched eyebrows, long philtrum, retrognathia, depressed nasal bridge, high arched palate, brachytelephalangy, brachymetatarsia, and mild scoliosis [Figure 1a-c]. He had dry, scaly skin lesions over the axilla, lateral side of the chest, and abdomen, suggestive of ichthyosis [Figure 1d-f]. On genitalia examination, he had a stretched penile length of 3.5 cm and a testicular volume of <2cc (Tanner staging – G1, no axillary and pubic hair). The rest of the systemic examination was normal, with no organomegaly. Fundus examination was normal. Based on the clinical presentation, the possibilities of multiple sulfatase deficiency and Sjogren–Larsson syndrome were considered.
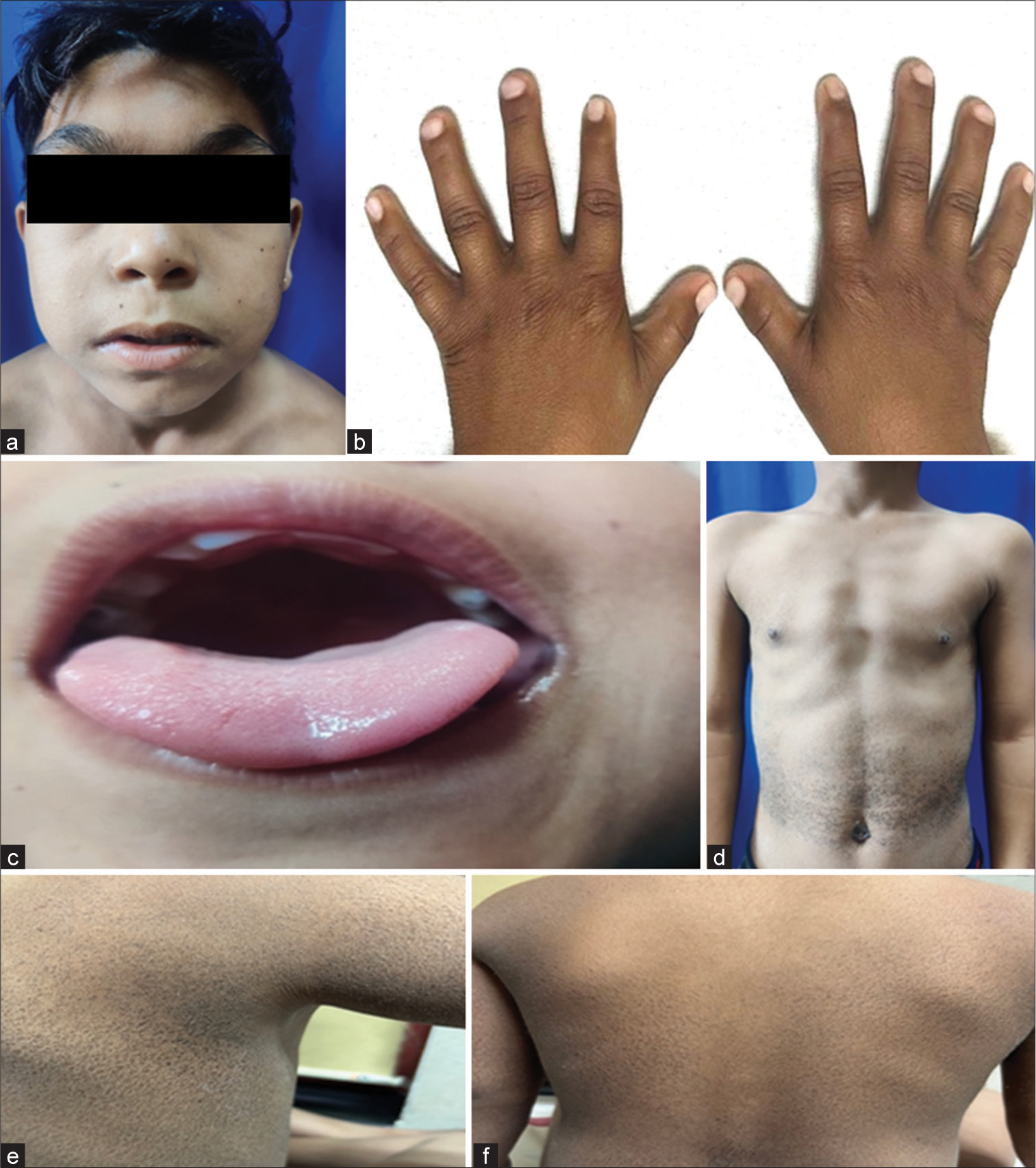
- Clinical features in the Index Child. (a-f) shows dysmorphic features and Ichthyosis. (d) shows pectus excavatum. (a) Facial dysmorphism, (b) brachytelephalangy, (c) high-arched palate, (d) ichthyosis over the abdomen, (e) ichthyosis over the axilla, (f) ichthyosis over the back.
He had a normal hemogram, renal, liver, and thyroid function tests, and a normal bone metabolic profile. He had normal cortisol levels (9.92 [3.09–16.66] μg/dL), low levels of testosterone (7.0 [241–827] ng/dL), luteinizing hormone (0.21 [2.8–6.8] mIU/mL), prolactin (50.4 [87– 392] mIU/L), and insulin growth factor 1 (9.05 [131– 490] ng/mL). The testing for smell with common odors revealed anosmia in the index child. The urinary testing for mucopolysaccharides was negative. Ultrasonogram study of the abdomen and pelvis was normal. The 2D echocardiography study was normal. The skeletal survey revealed abnormal development of distal phalanges of bilateral 1st – 4th fingers, coxa valga, and mild concavity with enlarged intervertebral discs in the lower thoracic and lumbar regions. Magnetic resonance imaging showed a normal pituitary gland, crowded posterior fossa with effacement of the fourth ventricle, and cerebellar tonsillar ectopia. Brain-evoked auditory response testing revealed bilateral moderate (60 dB) hearing loss. Clinical exome sequencing revealed a contiguous deletion of size (~8.10 Mb), spanning genomic location chrX:g.(_630898)_ (8732037_)del encompassing multiple genes including ANOS1, ARSL, NLGN4X, SHOX, STS was detected, suggestive of chromosome Xp22.3 deletion syndrome. The child is currently on a multidisciplinary follow-up.
LITERATURE SEARCH
A literature review was performed from the year 2023 to the oldest available report in English on Chromosome Xp22.3 deletion syndrome from the PubMed/MEDLINE, Google Scholar, and SCOPUS databases. All articles describing Chromosome Xp22.3 deletion syndrome were identified, and duplicates were removed. A total of 34 cases (excluding the index child) have been described in the literature and are summarized in Table 2.
| S. No | Author/year | Deletions | Age | Sex | Ich | SS | KS | CDP | MD | LWD | Epilepsy | ASD | ADHD | ID | OA | HL |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1. | Ballabio et al. 1988[20] | Xp22.3 | 6y | M | + | + | − | + | − | − | + | − | − | + | − | − |
| 2. | 6y | M | + | − | − | − | − | − | − | − | − | + | − | − | ||
| 3. | Bick et al. 1989[21] | Xp22.31 | 2.5m | M | + | − | + | + | − | − | − | − | − | − | − | − |
| 4. | Meindl et al. 1993[22] | Xp22.3 9.9 Mb | 9y | M | + | + | + | + | − | − | − | − | − | + | + | − |
| 5. | Schwinger et al. 1996[23] | Xp22.32 | 10y | F | − | + | − | − | − | − | − | − | − | − | − | − |
| 6. | Spranger et al. 1999[18] | Xp22.3 | 5y | M | − | + | − | − | + | + | + | − | + | − | − | + |
| 7. | Gohlke et al. 2000[24] | Xp22.3 | 8y | M* | + | − | + | − | − | − | + | − | − | + | − | − |
| 8. | 8y | M* | + | − | + | − | − | − | + | − | − | + | − | − | ||
| 9. | Doherty et al. 2003[1] | Xp22.3 | 24y | M | + | + | − | − | − | + | + | − | − | + | − | − |
| 10. | 22y | M | + | + | − | − | − | + | + | − | + | − | − | − | ||
| 11. | Boycott et al. 2003[19] | Xp22.3 6 Mb | 24y | M | − | − | − | − | − | − | + | − | + | + | − | − |
| 12. | 23y | M | − | + | − | − | + | − | − | − | + | − | − | − | ||
| 13. | Lonardo et al. 2007[9] | Xp22.3 5.5 Mb | 13y | M | + | + | − | + | − | − | − | − | + | + | − | + |
| 14. | Macarov et al. 2007[15] | Xp22.3 | 2y | M | + | − | + | − | − | − | − | − | − | − | − | − |
| 15. | 22y | M | + | − | + | − | − | − | − | − | − | − | − | − | ||
| 16. | 48y | M | + | − | + | − | − | − | − | − | − | + | − | − | ||
| 17. | Melichar et al. 2007[25] | Xp22.31 9.6 Mb | 1m | M | + | − | An | + | − | − | − | − | − | − | + | + |
| 18. | Puusepp et al. 2008[26] | Xp22.3 | 16y | F | − | + | − | − | + | − | + | + | + | + | − | − |
| 19. | Mochel et al. 2008[17] | Xp22.3 3.7 Mb | 17y | M | + | + | + | − | − | − | − | − | − | − | − | − |
| 20. | Van Steensel et al. 2008[4] | Xp22.3 8.4 Mb | 21y | M | + | + | − | − | − | − | + | − | − | + | − | − |
| 21. | Bukvic et al. 2010[8] | Xp23.33 7.7 Mb | 3y3m | M | + | − | − | − | + | − | − | − | + | − | − | − |
| 22. | 1y8m | F | − | − | − | − | − | − | − | − | − | − | − | − | ||
| 23. | Palka-Bayard-de-Volo et al. 2012[27] | Xp22.3 6.8 Mb | 11y | F | − | + | − | − | − | − | − | − | − | + | − | − |
| 24. | Khelifa et al. 2013[5] | Xp22.3 2 Mb | 14y | M | + | − | − | − | − | − | + | − | − | + | − | − |
| 25. | Xu et al. 2015[28] | Xp22.3 1.6 Mb | − | M | + | − | + | − | − | − | − | − | − | − | − | − |
| 26. | − | M | + | − | + | − | − | − | − | − | − | − | − | − | ||
| 27. | Vrečar et al. 2015[29] | Xp22.33 3 Kb | 9y | M | − | − | − | + | − | − | − | − | − | − | − | + |
| 28. | Berges-Raso et al. 2017[30] | Xp22.3 4.7 Mb | 39y | M | + | − | + | − | − | − | − | − | − | − | − | − |
| 29. | Amasdl et al. 2017[2] | Xp22.33 3.1Mb | 6y | M | − | + | − | − | − | − | − | − | − | − | − | + |
| 30. | 22y | M | − | + | − | − | − | − | − | − | − | − | − | + | ||
| 31. | Nagai et al. 2017[16] | Xp22.31 2.7 Mb | 6m | M | + | − | + | − | − | − | − | − | − | − | − | − |
| 32. | Sait et al. 2020[3] | Xp22.33 8.3 Mb | 20y | M | + | + | + | − | − | − | − | − | − | + | − | − |
| 33. | Ma et al. 2020[10] | Xp22.31 3.9 Mb | 14y | M | + | − | + | − | − | − | − | − | − | − | − | − |
| 34. | Xp22.3 5.8 Mb | 19y | M | + | − | + | − | − | − | − | − | − | − | − | − | |
| 35. | Index Study 2023 | Xp22.32 8.1 Mb | 10y | M | + | + | + | − | − | − | + | − | + | + | − | + |
An: Anticipated, F: Female, M: Male, m: months, y: years, *Monozygotic twins. ASD: Autism spectrum disorder, ADHD: Attention deficit hyperactivity disorder, CDP: Chondrodysplasia punctata, HL: Hearing loss, Ich: Ichthyosis, ID: Intellectual disability, KS: Kallmann syndrome, LWD: Leri-Weill dyschondrosteosis, MD: Madelung deformity, OA: Ocular albinism, SS: Short stature
DISCUSSION
The genetic analysis of the index child revealed a contiguous deletion of size (~8.10 Mb) in terminal Xp22.3 region encompassing multiple genes, including ANOS1 (KAL1), ARSL, NLGN4X, SHOX and STS, suggestive of chromosome Xp22.3 deletion syndrome [Figure 2]. This deletion was classified as a “likely pathogenic” copy number variation based on the American College of Medical Genetics guidelines.[7] The index child’s distinctive features were ichthyosis, global developmental delay, short stature, bilateral hearing loss, and hyperactivity. In the index child, along with signs of hypogonadism and anosmia, KS was diagnosed.
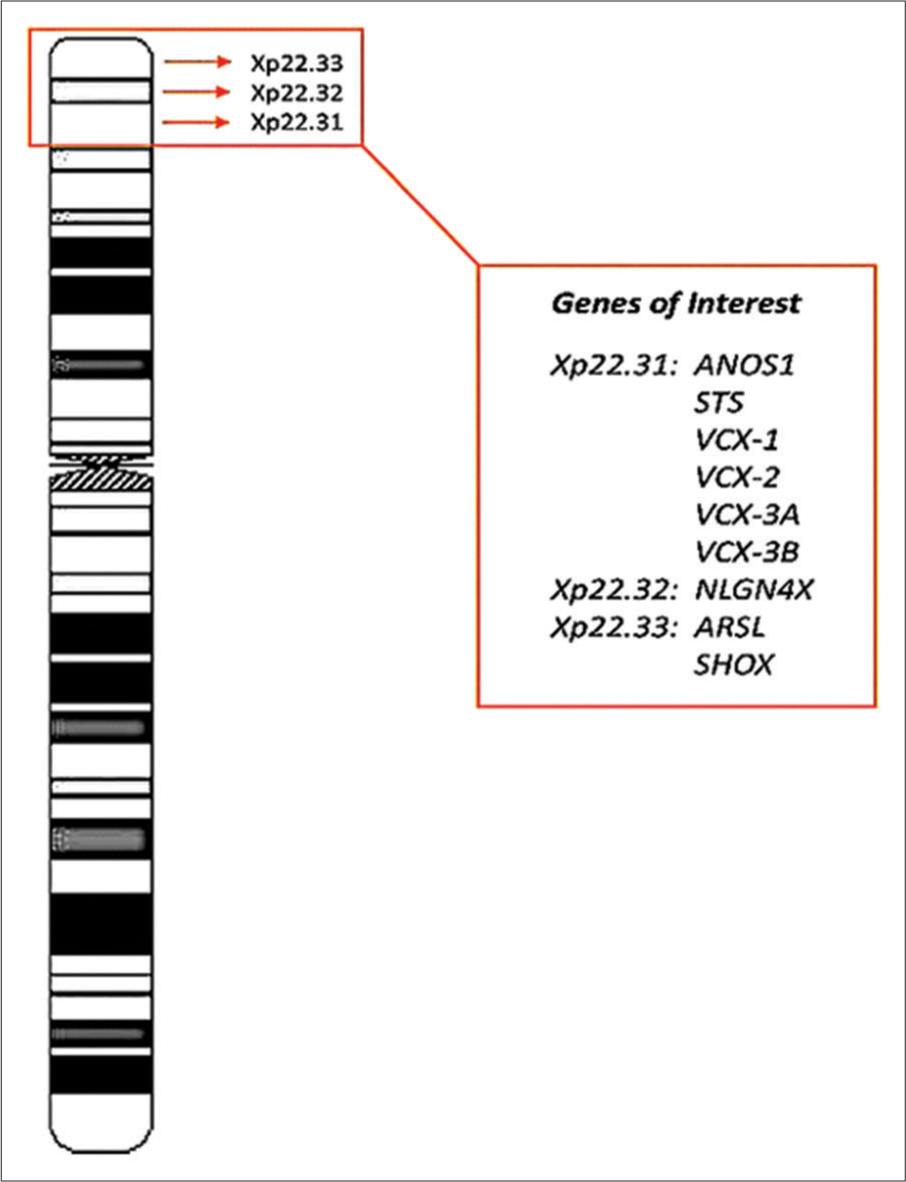
- Chromosome Xp22.3 region with associated significant Genes. G-banding Ideogram of X chromosome (800 bphs resolution) showing Xp22.3 region with the Genes of Interest.
The deletions of the Xp22.31 – p22.33 region result in contiguous gene syndromes with various phenotypical combinations of XLI, KS, short stature, CDP, developmental delay, ASD, ID, strabismus, obesity, ocular albinism, and bilateral hearing loss [Table 1].[1,8-10] The XLI (MIM#308100) is characterized by dry, dark-colored scales over the trunk and limbs, which is due to deletions of STS gene.[11] The absence of STS causes the accumulation of cholesterol sulfate in the stratum corneum, resulting in impaired skin permeability and hyperkeratosis.[12] The ANOS1 gene is located near the STS gene near the Xp22.3 locus, and the deletion of the ANOS1 gene causes X-linked KS. The ANOS1 gene encodes the anosmin-1 protein, which is present in the cerebral cortex and olfactory system. Ansomin-1 also mediates the adhesion and the axonal migration of gonadotrophin-releasing hormone (GnRH) neurons.[10] KS (MIM#308700) is due to deficiency of GnRH and hypoplasia of olfactory bulbs and is characterized by hypogonadotropic hypogonadism, anosmia/hyposmia, and other features may include cleft lip/palate, hearing loss, and unilateral renal agenesis.[10]
SHOX on the Xp gene is a homeobox gene involved in skeletal development and maturation. SHOX-related disorders have a highly variable phenotypic spectrum ranging from short stature to Leri-Weill dyschondrosteosis, which consists of a triad of mesomelia, short stature, and Madelung deformity (MD) (MIM#300582).[13] ARSL gene encodes ARSL, a member of the sulfatase family that is essential for the composition of bone and cartilage matrix.[14] The deletion of these genes results in short stature and X-linked chondrodysplasia punctata-1 (CDPX1). CDPX1 (MIM#302950) is characterized by brachytelephalangy – short distal phalanges, CDP – stippled epiphyses, nasomaxillary hypoplasia, short stature, hearing loss, vertebral abnormalities including dysplastic and hypoplastic vertebrae, developmental delay, ID, and cataracts.[14]
NLGN4X gene encodes the cell surface proteins of neurons. They are involved in forming and remodeling the synapses in the central nervous system. The deletion of NLGN4X causes X-linked ASD (MIM#300495) and X-linked ID (MIM#300495).[15-19] The deletion of other genes, including variably charged protein X-A (VCX-A), can also result in X-linked ID. The deletions of NLGN4X and VCX-A can cause variable phenotypes, including normal intellect.[5,15-17] Recently, in these associated contiguous gene syndromes involving Xp22.3, ADHD has been reported.[1,9,18,19]
In Xp22.3 microdeletions, cortical heterotopias, hypoplasia of corpus callosum, mild enlargement of sella turcica, olfactory bulb dysplasia (KS patients), and Dandy–Walker malformation have been reported.[4,8,10] Similarly, cerebellar tonsillar ectopia was noted in the index child, which has not been reported in the literature. The available literature on contiguous Xp22.3 deletion was reviewed and summarized in Table 2.
Limitations
The literature search included articles published only in the English language.
CONCLUSION
Contiguous gene syndromes exhibit complex disease phenotypes stemming from the microdeletion of multiple adjacent contiguous genes. These phenotypical associations serve as valuable indicators for the diagnosis, underscoring the critical role of recognizing them in establishing diagnoses and multimodal management planning for these patients.
Ethical approval
The Institutional Review Board approval is not required.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship
Nil.
References
- An Xp; Yq translocation causing a novel contiguous gene syndrome in brothers with generalized epilepsy, ichthyosis, and attention deficits. Epilepsia. 2003;44:1529-35.
- [CrossRef] [PubMed] [Google Scholar]
- Familial X/Y translocation encompassing ARSE in two moroccan siblings with sensorineural deafness. Cytogenet Genome Res. 2017;153:66-72.
- [CrossRef] [PubMed] [Google Scholar]
- Kallmann syndrome and X-linked ichthyosis caused by translocation between chromosomes X and Y: A case report. J Reprod Infertil. 2021;22:302-6.
- [CrossRef] [PubMed] [Google Scholar]
- Contiguous gene syndrome due to a maternally inherited 8.41 Mb distal deletion of chromosome band Xp22.3 in a boy with short stature, ichthyosis, epilepsy, mental retardation, cerebral cortical heterotopias and Dandy-Walker malformation. Am J Med Genet A. 2008;146A:2944-9.
- [CrossRef] [PubMed] [Google Scholar]
- Xp22.3 interstitial deletion: A recognizable chromosomal abnormality encompassing VCX3A and STS genes in a patient with X-linked ichthyosis and mental retardation. Gene. 2013;527:578-83.
- [CrossRef] [PubMed] [Google Scholar]
- Deletions and translocations involving the distal short arm of the human X chromosome: Review and hypotheses. Hum Mol Genet. 1992;1:221-7.
- [CrossRef] [PubMed] [Google Scholar]
- Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405-24.
- [CrossRef] [PubMed] [Google Scholar]
- Familial X;Y translocation with distinct phenotypic consequences: Characterization using FISH and array CGH. Am J Med Genet A. 2010;152A:1730-4.
- [CrossRef] [PubMed] [Google Scholar]
- Contiguous gene syndrome due to an interstitial deletion in Xp22.3 in a boy with ichthyosis, chondrodysplasia punctata, mental retardation and ADHD. Eur J Med Genet. 2007;50:301-8.
- [CrossRef] [PubMed] [Google Scholar]
- Novel microdeletion in the X chromosome leads to Kallmann syndrome, ichthyosis, obesity, and strabismus. Front Genet. 2020;11:596.
- [CrossRef] [PubMed] [Google Scholar]
- Inherited ichthyosis: Non-syndromic forms. J Dermatol. 2016;43:242-51.
- [CrossRef] [PubMed] [Google Scholar]
- X-Linked ichthyosis In: StatPearls. Treasure Island, FL: StatPearls Publishing; 2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK448149 [Last accessed on 2024 May 05]
- [Google Scholar]
- SHOX deficiency disorders In: Adam MP, Feldman F, Mirzaa GM, Pagon RA, Wallace SE, Bean LJ, eds. GeneReviews®. Seattle, WA: University of Washington, Seattle; 1993-2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1215 [Last accessed on 2024 May 05]
- [Google Scholar]
- Chondrodysplasia punctata 1, X-linked In: Braverman NE, Bober MB, Brunetti-Pierri N, Adam MP, Mirzaa GM, Pagon RA, eds. GeneReviews®. Seattle, WA: University of Washington, Seattle; 1993-2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1544 [Last accessed on 2024 May 05]
- [Google Scholar]
- Deletions of VCX-A and NLGN4: A variable phenotype including normal intellect. J Intellect Disabil Res. 2007;51(Pt 5):329-33.
- [CrossRef] [PubMed] [Google Scholar]
- Xp22.31 microdeletion due to microhomology-mediated break-induced replication in a boy with contiguous gene deletion syndrome. Cytogenet Genome Res. 2017;151:1-4.
- [CrossRef] [PubMed] [Google Scholar]
- Normal intelligence and social interactions in a male patient despite the deletion of NLGN4X and the VCX genes. Eur J Med Genet. 2008;51:68-73.
- [CrossRef] [PubMed] [Google Scholar]
- Léri-Weill syndrome as part of a contiguous gene syndrome at Xp22.3. Am J Med Genet. 1999;83:367-71.
- [CrossRef] [Google Scholar]
- A familial contiguous gene deletion syndrome at Xp22.3 characterized by severe learning disabilities and ADHD. Am J Med Genet A. 2003;122A:139-47.
- [CrossRef] [PubMed] [Google Scholar]
- X/Y translocation in a family with X-linked ichthyosis, chondrodysplasia punctata, and mental retardation: DNA analysis reveals deletion of the steroid sulphatase gene and translocation of its Y pseudogene. Clin Genet. 1988;34:31-7.
- [CrossRef] [PubMed] [Google Scholar]
- Male infant with ichthyosis, Kallmann syndrome, chondrodysplasia punctata, and an Xp chromosome deletion. Am J Med Genet. 1989;33:100-7.
- [CrossRef] [PubMed] [Google Scholar]
- Analysis of a terminal Xp22.3 deletion in a patient with six monogenic disorders: Implications for the mapping of X linked ocular albinism. J Med Genet. 1993;30:838-42.
- [CrossRef] [PubMed] [Google Scholar]
- Short stature in a mother and daughter with terminal deletion of Xp22.3. Am J Med Genet. 1996;63:239-42.
- [CrossRef] [Google Scholar]
- Interstitial deletion in Xp22.3 is associated with X linked ichthyosis, mental retardation, and epilepsy. J Med Genet. 2000;37:600-2.
- [CrossRef] [PubMed] [Google Scholar]
- A male infant with a 9.6 Mb terminal Xp deletion including the OA1 locus: Limit of viability of Xp deletions in males. Am J Med Genet A. 2007;143A:135-41.
- [CrossRef] [PubMed] [Google Scholar]
- Girl with partial Turner syndrome and absence epilepsy. Pediatr Neurol. 2008;38:289-92.
- [CrossRef] [PubMed] [Google Scholar]
- Array-CGH characterization of a de novo t(X;Y)(p22;q11) in a female with short stature and mental retardation. Gene. 2012;504:107-10.
- [CrossRef] [PubMed] [Google Scholar]
- Novel homozygous deletion of segmental KAL1 and entire STS cause Kallmann syndrome and X-linked ichthyosis in a Chinese family. Andrologia. 2015;47:1160-5.
- [CrossRef] [PubMed] [Google Scholar]
- Brachytelephalangic chondrodysplasia punctata caused by new small hemizygous deletion in a boy presenting with hearing loss. Mol Cytogenet. 2015;8:83.
- [CrossRef] [PubMed] [Google Scholar]
- Kallmann syndrome and ichthyosis: A case of contiguous gene deletion syndrome. Endocrinol Diabetes Metab Case Rep. 2017;2017:EDM170083.
- [CrossRef] [PubMed] [Google Scholar]




