
Translate this page into:
Chiari Type I malformation yielded to the diagnosis of Crouzon syndrome
Address for correspondence: Dr. Mehmet Osman Akcakaya, Taksim Training and Research Hospital, Department of Neurosurgery, Sıraselviler Cad. No: 112, Beyoglu 34433, Istanbul, Turkey. E-mail: moakcakaya@gmail.com
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Chiari malformation Type I (CM-I) related to syndromic craniosynostosis in pediatric patients has been well-studied. The surgical management consists of cranial vault remodeling with or without posterior fossa decompression. There were also cases, in whom CM-I was diagnosed prior to the craniosynostosis in early childhood. We present a 16-year-old boy who admitted with symptoms related to CM-I. With careful examination and further genetic investigations, a diagnosis of Crouzon syndrome was made, of which the patient and his family was unaware before. The patient underwent surgery for posterior fossa decompression and followed-up for Crouzon's syndrome. To our knowledge, this is the only case report indicating a late adolescent diagnosis of Crouzon syndrome through clinical symptoms of an associated CM-I.
Keywords
Chiari malformation Type I
craniosynostosis
Crouzon syndrome
posterior fossa decompression

Introduction
Crouzon syndrome is an autosomal dominant disorder with high penetrance. The syndrome is named after French neurosurgeon Octave Crouzon, who described this rare genetic disorder first time in 1912.[1] Although it is encountered rarely, Crouzon syndrome constitutes almost 5% of all craniosynostoses with an approximate birth prevalance of 1/25,000-1/50,000.[23] It is the most frequent form of syndromic craniosynostoses. The syndrome is characterized by abnormal head shape, midfacial hypoplasia, maxillary hypoplasia, mandibular prognathism, ocular hypertelorism, proptosis, and airway obstruction due to premature fusion of multiple calvarial and skull base sutures within the first year of life.[34] However, the clinical picture may vary greatly from mild to severe midfacial and orbital anomalies.
The relationship between craniosynostosis and Chiari malformation Type I (CM-I) has been well-documented. CM-I has a tendency to accompany syndromic craniosynostosis more commonly than sporadic synostosis.[5] The incidence of CM-I in Crouzon syndrome is about 70%.[6] Herein, we present a 16-year-old boy who admitted with symptoms related to CM-I and underwent suboccipital decompression. However, on physical examination his cruzonoid features drawed attention. Further genetic investigations yielded to the diagnosis of Crouzon syndrome.
Case Report
A 16-year-old, formerly healthy boy admitted to the outpatient clinic with occasional headache and neck pain. On physical examination it was remarked that he had characteristic features of syndromic craniosynostoses: Hypertelorism, proptosis, midfacial hypoplasia, and abnormal head shape [Figures 1a and b]. However, the patient and his family did not admit to a hospital for this reason before, despite prominent Cruzonoid features. The patients’ neurological examination revealed no abnormalities including pinprick, touch, pain, and temperature sensations in both upper extremities. Anteroposterior (AP) and lateral plain radiographs of head and a cranial computed tomography (CT) demonstrated midfacial and orbital hypoplasia, the fusion of bilateral coronal and lambdoid sutures and sagittal suture, alongside with the increase of AP diameter of the head [Figures 2a and b]. The patient did not have a history of delayed maturation. His psychomotor development was completely normal. The patient did not suffer from any systemic disorders including chronic hypertension. Fundoscopic examination showed no papilledema. Cranial and spinal magnetic resonance imaging (MRI) studies were obtained and it showed a 18 mm cerebellar tonsil herniation into the foramen magnum with accompanying syringomyelia between Th 4 and Th 7 [Figure 3]. Surgery was planned in order to decompress the posterior cranial fossa. Using a median incision, a suboccipital craniectomy, total C1 arcusectomy and partial C2 laminectomy was performed. The dura was opened in Y-form and after arachnoid dissection duraplasty was performed. There were no significant events in the postoperative course and the patient was discharged from the hospital with no neurological deficits. The patient was then referred to the genetics department for the further evaluation of the craniosynostosis. Phenotypical features like the typical dismorphic facies, ocular proptozis and hypertelorism, parrot-like nose, and frontal bossing alongside with the patients’ mothers’ history of four recurrent intrauterine fetal losses supported the diagnosis of Crouzon syndrome in our patient. Regular follow-up examinations were scheduled for the observation of orbital deformities and vision. Control MRI showed complete resolution of the tonsillar herniation and significant reducement of the syringomyelia both in length and thickness [Figures 4a and b]. Four years after the operation the patient is still doing well with no neurological or ocular deficits and with the relief of his symptoms at the admission.
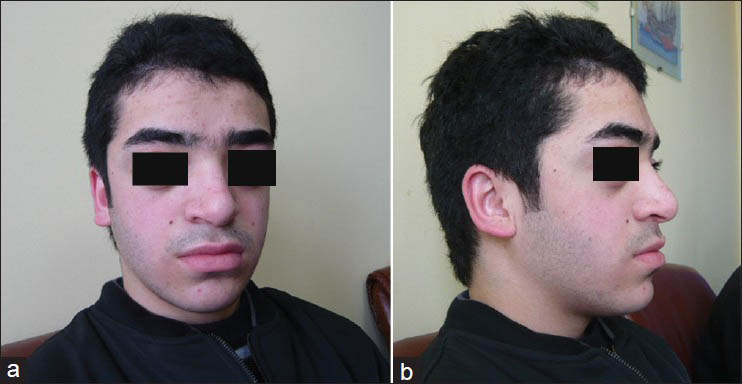
- (a and b) Photographs show characteristic features of syndromic craniosynostoses in our patient: Hypertelorism, proptosis, midfacial hypoplasia, and abnormal head shape. (Published with permission and informed consent of the patient)
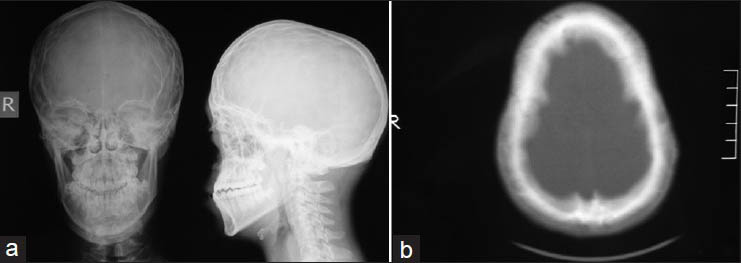
- (a) Anteroposterior (AP) and lateral plain radiographs of head shows increase of AP diameter of the head and midfacial-orbital hypoplasia. Fusion of multiple calvarial sutures is also remerkable, (b) Axial cranial computed tomography scan demonstrated the abnormal head shape and the fusion of bilateral coronal and lambdoid sutures alongside with sagittal suture
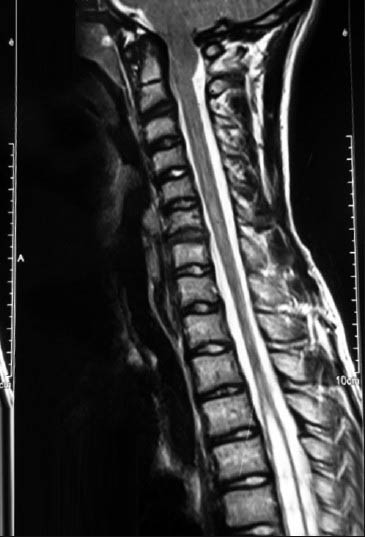
- Sagittal T2-weighted magnetic resonance imaging shows a 18 mm cerebellar tonsil herniation into the foramen magnum with accompanying syringomyelia between Th 4 and Th 7 with the largest thickness of 11 mm
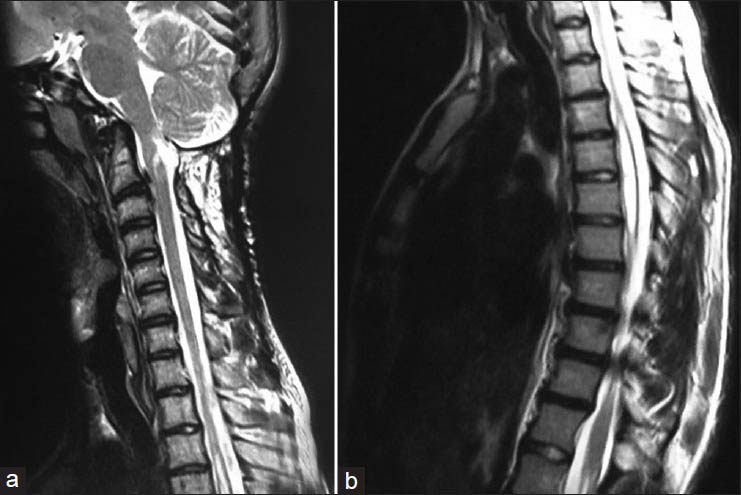
- (a) Sagittal T2-weighted MRI of the craniocervical junction showed complete resolution of 18 mm cerebellar tonsil herniation, (b) Sagittal T2-weighted thoracal MRI showed relative resolution of the accompanying syringomyelia between Th 4 and Th 7 with the largest thickness of 7 mm
Discussion
The association between syndromic craniosynostosis and CM-I has been well established. The premature fusion of cerebral sutures has been postulated as the mechanism leading to the development of CM-I in patients with syndromic craniosynostosis. Especially premature fusion of the lambdoid suture has been accepted as a crucial developmental anomaly, which results in a relatively small posterior fossa.[7] Cinalli et al., have reported that CM-I is present in 70% of patients with Crouzon syndrome. On contrary, CM-I was found only in 1.9% of patients with Apert syndrome.[78] They have proposed, that this relationship is due to earlier closure of sagittal and lambdoid sutures in Crouzon syndrome compared to Apert syndrome.[7] Hydrocephalus, jugular venous stenosis leads to venous hypertension and associated brain malformations have been postulated as the other mechanisms leading to CM-I development.[9]
Current knowledge about the genetics of syndromic craniosynostoses have been expanded with recent studies. Mutations in three of four fibroblast growth factor receptor (FGFR) genes have been demonstrated to be responsible for various types of syndromic craniosynostoses including Crouzon syndrome.[3] The mutations in Crouzon syndrome are related with several mutations in FGFR2.[3] Fujisawa et al., have demonstrated that a missense mutation in FGFR2 gene (Tyr281Cys) is responsible for the development of CM-I in patients with Crouzon syndrome.[3]
The current case is an unique example of Crouzon syndrome. Despite prominent outlook, the patient or his family was never attempted to seek for professional medical help. The syndrome was only diagnosed when the patient was admitted to our department with symptoms of CM-I. Strahle et al., presented a series of patients with CM-I associated with craniosynostosis.[9] In their series of 29 patients, 17 patients had CM-I diagnosed prior to craniosynostosis.[9] However, the mean age of the whole patient group was 1.8 years (range 2 months-9 years). This data shows that both craniosynostosis or craniosynostosis related CM-I are expected to be diagnosed in early childhood. CM-I could be diagnosed before the craniosynostosis in some instances, but the age of our patient at diagnosis was 16. To our knowledge, this is the only case report indicating a late adolescent diagnosis of Crouzon syndrome through clinical symptoms of an associated CM-I.
Surgical approach to craniosynostosis related CM-I may include cranial vault remodeling with an adequate posterior fossa decompression.[910] However, most neurosurgeons agree with the conservative follow-up of patients with CM-I unless it is not symptomatic or associated with spinal syringomyelia.[9] Strahle et al., reported some of his patients’ CM-I was resolved or regressed with cranial vault remodeling only, without posterior fossa decompression.[9] However, these are only pediatric cases in early childhood. For our patient, we do not consider cranial vault remodeling as an option, instead we applied a classical posterior fossa decompression for CM-I. Strahle et al., underlined the risk of venous bleeding due to abnormal venous sinuses and increased venous hypertension, therefore suggested a posterior fossa decompression without dural opening and C1 arcusectomy. We did not encounter any venous bleeding during the surgery, where we opened dura and performed a duraplasty.
Conclusion
Pediatric patients with CM-I should be carefully examined for the clinical signs and features of Crouzon syndrome or other syndromic craniosynostosis. In mild clinical forms or in case of a late diagnosis, posterior fossa decompression without cranial remodeling should be kept in mind as a treatment option.
Source of Support: Nil.
Conflict of Interest: None declared.
References
- Dysostose cranio-faciale hereditaire (Hereditary cranio-facial dysostose) Bulletin de la Societe des Medecins des Hopitaux Paris. 1912;33:545-55.
- [Google Scholar]
- A novel fibroblast growth factor receptor 2 mutation in Crouzon syndrome associated with Chiari Type I malformation and syringomyelia. J Neurosurg. 2002;97:396-400.
- [Google Scholar]
- Early orthodontic management of Crouzon syndrome: A case report. J Maxillofac Oral Surg. 2009;8:74-6.
- [Google Scholar]
- The incidence of Chiari malformation in nonsyndromic, single suture craniosynostosis. Childs Nerve Syst. 2010;26:771-4.
- [Google Scholar]
- Central sleep apnea and associated Chiari malformation in children with syndromic craniosynostosis: Treatment and outcome data from a supraregional national craniofacial center. J Neurosurg Pediatr. 2013;11:296-301.
- [Google Scholar]
- Chronic tonsiller herniation in Crouzon's and Apert's syndromes: The role of premature synostosis of the lambdoid suture. J Neurosurg. 1995;83:575-82.
- [Google Scholar]
- Occipital remodelling and suboccipital decompression in severe craniosynostosis associated with tonsiller herniation. Neurosurgery. 1998;42:66-71.
- [Google Scholar]
- Resolution of syndromic craniosynostosis-associated Chiari malformation Type I without suboccipital decompression after posterior cranial vault release. J Neurosurg Pediatr. 2012;9:111-5.
- [Google Scholar]




