
Translate this page into:
Afebrile Seizures as Initial Symptom of Hypocalcemia Secondary to Hypoparathyroidism
Address for correspondence: Dr. Anastasia Gkampeta, St. Kyriakidi 1, Thessaloniki 54636, Greece. E-mail: anastagab@yahoo.gr
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Hypocalcemia is rare in childhood and caused, among other conditions, by hypoparathyroidism. DiGeorge syndrome is the most common cause of hypoparathyroidism in childhood. Presentation of a rare cause of hypocalcemia in childhood and the necessity of measuring serum electrolyte levels in patients presenting with afebrile seizures. a 7.5-year-old female child presented with afebrile seizures lasting 5 min with postictal drowsiness. A similar episode 1 month ago is described. On admission, a positive Trousseau sign, papilledema, and long QTc on electrocardiography were detected. Laboratory testing revealed hypocalcemia, increased creatine phosphokinase and phosphate levels, decreased levels of parathormone, with normal thyroid function and normal levels of blood gases. considering the diagnosis of hypoparathyroidism possible, we started on calcium gluconate solution 5% intravenously and calcium carbonate per os. 48 h later, the child transferred to tertiary hospital for further evaluation. The laboratory findings revealed 25-OH Vitamin D deficiency with normal cortisol levels and the absence of autoantibodies. Kidney and brain imaging and also the electroencephalogram were normal. Calcium carbonate, magnesium, and Vitamin D were administered per os. The child discharged from hospital with complete resolution of symptoms. Since then, she is in treatment with calcium carbonate and Vitamin D per os. Hypoparathyroidism is rare in childhood. We underline the necessity of measuring serum electrolyte levels in patients presenting with afebrile seizures.
Keywords
Afebrile seizures
children
hypocalcemia
hypoparathyroidism

INTRODUCTION
Hypoparathyroidism is rare in childhood, characterized by inadequate parathyroid hormone (PTH), resulting in hypocalcemia and hyperphosphatemia and also in some cases, calciuria and 25-OH Vitamin D deficiency.[12] Hypoparathyroidism can be transient, inherited, or acquired, caused by inability to synthesize or secrete PTH, abnormal parathyroid gland development, destruction of parathyroid tissue, or peripheral resistance to PTH. The most common cause of congenital hypoparathyroidism is DiGeorge syndrome.
Signs and symptoms of hypoparathyroidism vary, with main symptoms related to hypocalcemia. Chronic hypocalcemia may be asymptomatic. However, acute hypocalcemia usually results in severe symptoms including laryngospasm, stridor, airway obstruction, neuromuscular irritability, cognitive impairment, prolonged QT interval, and cardiac arrhythmias. Neuromuscular manifestations may include circumoral numbness, paresthesia, hyperreflexia, muscle cramping, tetany, and seizures.[2] Hyperreflexia is manifested by carpal spasms (Trousseau sign) when arterial blood flow to the hand is occluded with a blood pressure cuff inflated above the systolic blood pressure for 3–5 min and ipsilateral facial spasms (Chvostek sign) with tapping of the facial nerve just in front of the ear.
The current paper presents a case of a 7.5-year-old female child presented with afebrile seizures as initial symptom of hypocalcemia secondary to hypoparathyroidism.
CASE REPORT
A 7.5-year-old female child was referred to our clinic due to an episode of afebrile generalized tonic-clonic seizures lasting 5 min, followed by postictal drowsiness that lasted 3 h. In the past, the child had a similar episode 1 month ago. According to the parents, there was no history of recent head injury or infectious disease. The child had an unremarkable perinatal and medical history (she was born full-term, weighing 3.100 g, with Apgar score 110 and 510). Growth and development were normal until now. In her family history, her mother and mother's sister were suffering from collagen disorders.
On admission, a positive Trousseau sign, papilledema, and long QTc on electrocardiography were detected (QTc 0.53 ms, normal ≤0.44) [Figure 1]. Physical and neurological examinations were unremarkable and vitals of the child were stable. Her complete blood count and arterial blood gases were normal (PCO2: 35 mmHg, PO2: 100 mmHg, pH: 7.45, white blood cell: 6.16 × 103/μL, neutrophils: 75%, lymphocytes: 14.3%, red blood cell: 4.44 × 106/μL, hemoglobin B: 12.6 g/dl, hematocrit: 35.1%, platelet: 175 × 103/μL). Biochemical values revealed hypocalcemia (Ca 4, mg/dl, normal limits: 8.4–10.2 mg/dl), slightly low serum magnesium values (1.6 mg/dl, normal limits: 1.7–2.6 mg/dl), increased creatine phosphokinase (CPK) values (1790 IU/l or 30.43 µkat/L, normal limits: 20–180 IU/l or 0–2.16 µkat/L), and increased phosphate values (8 mg/dl, normal limits: 3.4–6.2 mg/dl). Based on the report of the investigations, hypoparathyroidism was suspected; hence, we started on calcium gluconate solution 5% intravenously (16 ml calcium gluconate 5%, which contain 800 mg calcium gluconate) and calcium carbonate per os (1000 mg 3 times daily). 48 h later, the child transferred to tertiary hospital for further evaluation. Further laboratory findings revealed very low PTH values (5 pg/ml or 0.75 pmol/L, normal: 10–65 pg/ml or 1.2–5.8 pmol/L), 25-OH Vitamin D deficiency with normal thyroid function, normal cortisol values, and the absence of antithyroid peroxidase antibodies. CPK values were measured and found within normal limits, excluding the possibility of any muscular disease. Kidney and brain magnetic resonance imaging, echocardiogram, and electroencephalogram were all normal. The absence of cardiac defects, absence of distinct facial features, and unremarkable medical history during neonatal period helped us exclude the diagnosis of DiGeorge syndrome.
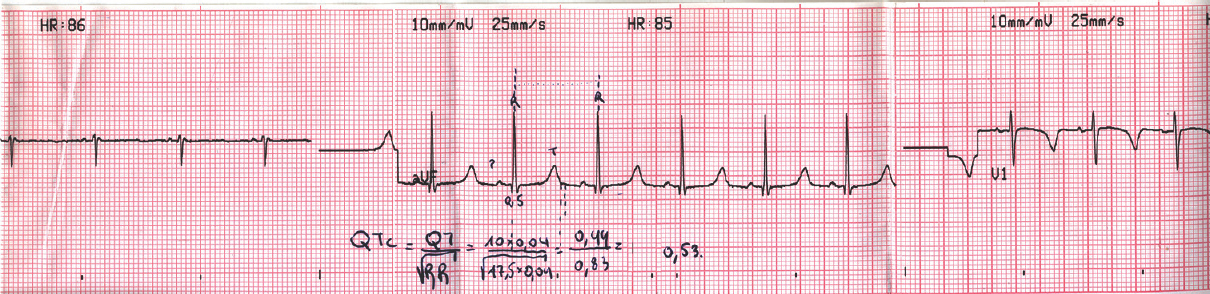
- Long QTc on electrocardiography of a 7.5-year-old female child presented with afebrile seizures as initial symptom of hypocalcemia secondary to hypoparathyroidism (QTc 0.53 ms, normal ≤0.44)
Calcium carbonate (1000 mg 3 times daily), magnesium (100 mg daily), and Vitamin D (0.25 mcg daily) were administered per os. The child discharged from hospital with complete resolution of symptoms. Since then, she is in treatment with calcium carbonate (1000 mg 3 times daily) and Vitamin D (0.25 mcg daily) per os.
DISCUSSION
Hypoparathyroidism is a rare disease in childhood, characterized by inadequate PTH, resulting in hypocalcemia and hyperphosphatemia.[3] Management of hypoparathyroidism is complex, depending on underlying etiology and is individualized, depending on the severity of hypocalcemia. Treatment goals include control of hypocalcemic symptoms, with serum calcium and phosphate in the low normal range. Conventional therapy includes calcium carbonate and active Vitamin D. Acute and symptomatic hypocalcemia need to be corrected with intravenous bolus or continuous infusion of calcium through a central venous catheter. Maintenance therapy with oral calcium replacement is given several times daily to increase the serum calcium level, using calcium citrate, calcium glubionate, or calcium carbonate. Chronic treatment of hypoparathyroidism may be difficult to manage due to the need for a sensitive balance between calcium and phosphate levels to prevent nephrolithiasis, nephrocalcinosis, and soft tissue calcification in the kidney.[4]
A daily subcutaneous injection of recombinant human PTH was recently approved by Food and Drug Administration as therapy in certain adults whose calcium levels cannot be controlled on calcium supplementation and active Vitamin D. There is a lack of evidenced-based studies concerning the safety and efficacy of recombinant human PTH in children with severe hypoparathyroidism who are refractory to conventional therapy.[5]
Hypocalcemia is a rare cause of afebrile seizures in childhood. We describe a patient with hypocalcemia due to hypoparathyroidism. Reports of children presenting with afebrile seizures secondary to idiopathic hypoparathyroidism are uncommon in literature. However, there are many reports in literature concerning children with hypoparathyroidism whose delayed diagnosis resulted in intractable seizures, mental retardation, and calcification of brain areas.[6] Eaton et al. in 1939 first reported the idiopathic hypoparathyroidism as causative factor for basal ganglia calcification.[7] In 1989, Mithal et al. reported the clinical, biochemical, and radiological findings in 13 patients diagnosed with hypoparathyroidism (mean age: 9 years). Nine patients presented with a history of generalized seizures and two were in acute hypocalcemic crisis at the time of admission. Nine patients underwent a head computed tomography scan and five patients had evidence of basal ganglia calcification.[8] According to literature, basal ganglia calcification occurs in 73.8% of patients with idiopathic hypoparathyroidism and correlates with the duration and the severity of hypocalcemia.[7] The pathogenesis of basal ganglia calcification in patients diagnosed with idiopathic hypoparathyroidism is yet not well understood. However, early diagnosis and treatment of hypocalcemia reduces the progression of basal ganglia calcification.
This case report highlights the importance of early diagnosis of children with hypoparathyroidism and the necessity of measuring serum electrolyte levels in patients presenting with afebrile seizures. We also highlight the presence of papilledema as a rare but recognized complication of hypocalcaemia secondary to hypoparathyroidism.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- Hypoparathyroidism-disease update and emerging treatments. Ann Endocrinol (Paris). 2015;76:84-8.
- [Google Scholar]
- Parathyroid hormone therapy for hypoparathyroidism. Best Pract Res Clin Endocrinol Metab. 2015;29:47-55.
- [Google Scholar]
- Teriparatide (rhPTH) treatment in children with syndromic hypoparathyroidism. J Pediatr Endocrinol Metab. 2014;27:53-9.
- [Google Scholar]
- Recurrent seizures, mental retardation and extensive brain calcinosis related to delayed diagnosis of hypoparathyroidism in an adolescent boy. J Clin Neurosci. 2015;22:894-6.
- [Google Scholar]
- Symmetric cerebral calcifications, particularly of the basal ganglia demonstrate roentgenographically; calcification of the cerebral blood vessels. Arch Neurol Psychiatry. 1939;41:921-42.
- [Google Scholar]
- Spontaneous hypoparathyroidism: Clinical, biochemical and radiological features. Indian J Pediatr. 1989;56:267-72.
- [Google Scholar]




