
Translate this page into:
Primary intradural extramedullary Ewing sarcoma: Review of literature and update for a standard protocol
*Corresponding author: Alberto Morello, Department of Neuroscience, Neurosurgery Unit, Città della Salute e della Scienza di Torino, University Hospital, University of Turin, Turin, Italy. alberto.morello@unito.it
-
Received: ,
Accepted: ,
How to cite this article: Da Rin Vidal T, Morello A, Impalà G, Meyer A, Lanotte M, Bertero L, et al. Primary intradural extramedullary Ewing sarcoma: Case report and Review of literature and update for a standard protocol. J Neurosci Rural Pract. doi: 10.25259/JNRP_289_2024
Abstract
Ewing sarcoma (ES) is a rare, undifferentiated, and malignant mesenchymal tumor primarily affecting children and young adults. It typically presents as a lytic bone lesion located in the diaphysis of long bones or the flat bones of the pelvis, with the most common sites of metastasis being the lungs, skeletal system, and bone marrow. Primary intradural extramedullary Ewing sarcoma (IEES) is extremely rare, and its clinical presentation often overlaps with that of other spinal tumors, which can complicate diagnosis and treatment. We report a case of a young male patient, who was admitted with primary intradural extramedullary ES in the lumbar region. Magnetic resonance imaging revealed a large intradural, extramedullary mass extending from L2 to L5, with moderate contrast enhancement, initially suggesting a diagnosis of ependymoma. Consequently, the patient underwent an L2–L5 laminotomy with partial resection of the tumor. Histopathological and immunohistochemical analyses confirmed the diagnosis of IEES. After a multidisciplinary collegial evaluation of the case, the patient underwent adjuvant treatment with systemic chemotherapy. IEES is a rare condition, but it still merits consideration as a differential diagnosis of spinal tumors. Despite advances in treatment modalities, the literature review underscores the risk of local recurrence and distant metastasis, drawing attention to the importance of ideally pursuing radical surgery and effective oncologic treatment.
Keywords
Ewing sarcoma
Extra-skeletal
Intradural
Mesenchymal
Metastases
Spinal tumor

INTRODUCTION
Ewing sarcoma (ES) is a rare, undifferentiated, and malignant mesenchymal tumor originating from the neuroectoderm, primarily affecting children and adolescents. In most cases, ES manifests as a lytic bone lesion, typically located in the diaphysis of long bones or the flat bones of the pelvis. The most common sites of metastasis are the lungs, skeletal system, and bone marrow. According to the 2020 World Health Organization Classification of Soft-Tissue Tumors, ES is categorized between undifferentiated small round cell sarcoma of bone and soft tissues and presents EWSR1 gene fusion which distinguishes it from the other subsets of the group, all differing from a clinical, pathological, and molecular point of view.[1] Extraskeletal ES subtype was first described in 1969 and usually presents as a mass in deep soft tissues with local pain involving the lower extremities, paravertebral region and chest wall, less commonly upper limb, head, neck, pelvis, and the retroperitoneal region.[2] Compared to classical ES, the distribution is bimodal, with peaks evident both below 5 years and above 35 years of age, and shows no sex predominance. Moreover, primary intradural extramedullary ES (IEES) is an extremely rare spinal cord tumor, and the resemblance of its presentation to other spinal tumors, such as schwannomas, meningiomas, and ependymomas, further complicates its identification and management. It, therefore, poses unique diagnostic and therapeutic challenges.
Here, we present the case of a young male patient with primary IESS in the lumbar region, who was admitted to Centro Traumatologico Ortopedico of Città della Salute e della Scienza (Turin, Italy) in November 2023. This case is accompanied by a review of the existing literature and the proposal of a flowchart outlining a standard protocol for the diagnosis and management of such cases.
CASE REPORT
A 36-year-old male presented with worsening lower back pain, radiating down the lower right extremity, which had been progressively increasing over the course of one year despite poor response to pain relief therapy with nonsteroidal anti-inflammatory drugs. He later developed burning paresthesia, gait difficulty due to dorsiflexion deficiency of the right foot, and a feeling of incomplete defecation and urination with persistence of urge after voiding.
Lumbar magnetic resonance imaging (MRI) revealed a large intradural extramedullary mass in the cauda equina, extending from the lower endplate of L2 to the upper endplate of L5, with a vertical length of 7.5 cm, completely filling the spinal canal [Figure 1]. The mass had a solid appearance and showed hypointensity on T1-weighted images, isointensity on T2-weighted images, and a modest, slightly inhomogeneous, contrast enhancement, suggesting the diagnosis of a probable myxopapillary ependymoma.
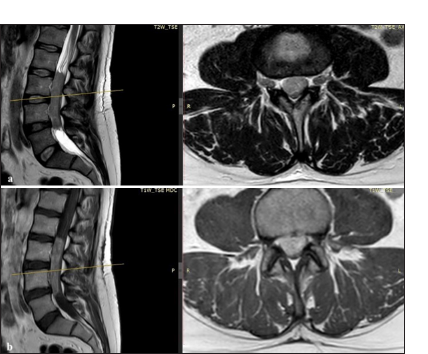
- (a) Sagittal and axial lumbar MRI images. T2 non-contrasted sequence showing low T2 signal-intensity intradural/extramedullary lesion extending from the lower aspect of L2 to L4–L5 disc level. (b) Sagittal and axial T1 contrasted image showing diffuse homogenous enhancement of the lesion. L: Lesion, T1W: T1-weighted MRI image without contrast, T2W: T2-weighted MRI image without contrast, TSE: Turbo spin echo, MDC: Mezzo di contrasto (contrast agent), AX: Axial, P: Posterior, R: Right, L: Left.
Neurological physical examination revealed significant sthenic deficiency in the lower right limb (Medical Research Council [MRC] 4/5 at thigh flexion and leg extension, MRC 3/5 at foot dorsiflexion, and MRC 2/5 at foot plantar flexion) with hypoelicitable patellar osteotendinous reflex associated. Signs of upper motor neuron suffering were present in the left lower limb (lively osteotendinous reflexes with reflexogenic area extension).
During hospitalization, an MRI of the whole spine was also performed, which showed no further expansive lesions at the endocanal level.
The patient was referred to the neurosurgery department and underwent an L2–L5 laminotomy. Due to the extensive involvement of nerve roots surrounding the tumor and the decreased amplitude observed in intraoperative neurological monitoring, only a partial resection of the tumor was performed. During the surgery, a reduction in the amplitude of both motor-evoked potentials and somatosensory-evoked potentials was recorded, particularly in the distal part of the right lower limb.
Pain and paresthesia in lower limbs disappeared after the surgery and the patient recovered completely from the proximal muscle weakness of the right lower limb. On the other hand, right foot dorsi- and flexion did not improve (MRC 1/5). In addition, weaning from the bladder catheter proved impossible due to persistent urinary retention.
Histological examination of hematoxylin and eosin slides showed fragments of dense connective tissue populated by a proliferation of undifferentiated small round cells with irregular nuclei, high nucleus-to-cytoplasm ratio, sparse chromatin, and either absent or clear cytoplasm [Figure 2]. Rare mitotic figures were present as well (2/10 High-Power Field).
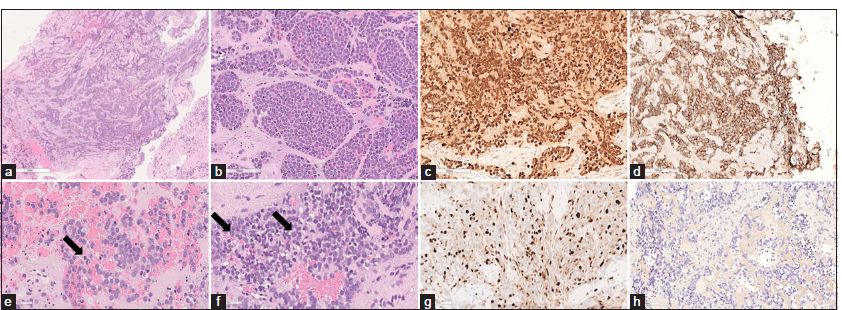
- Photomicrographs of the resected specimen. (a-d) Hematoxylin and eosin stain shows a dense proliferation of small round cells with scant to absent cytoplasm, high nucleus/cytoplasm ratio with fine strippled chromatin, (c, arrow) sparse mitoses and (d, arrow) small nucleoli. (e) Immunohistochemical reactions demonstrating diffuse nuclear positivity for NKX2.2, (f) membranous positivity for CD99 and (g) a proliferation index of 40%. (h) Staining for cytokeratins AE1/AE3 was negative.
Immunohistochemical reactions showed diffuse membrane positivity for CD99 and nuclear positivity for NKX2.2. INI1 expression was retained. Stainings for cytokeratins AE1/AE3, desmin, S100, GFAP, CD34, chromogranin A, synaptophysin, Wilms tumor Gene 1 (WT1), leukocyte common antigen (LCA), CD3, and CD20 were negative.
After diagnosis, the patient underwent a positron emission tomography (PET)-computed tomography (CT), which ruled out the dissemination of the disease. After a multidisciplinary collegial evaluation of the case, the patient underwent adjuvant treatment with systemic chemotherapy (ChT) (including doxorubicin + cyclophosphamide), and any radiotherapy (RT) was postponed until the reduction of the tumor mass with the intention of preserving the enveloped nerve roots as much as possible. At this time, the patient received three cycles of ChT without complications.
DISCUSSION
IEES is an extremely rare pathology, with very few cases reported to date. Table 1 shows a summary of them.[3-33] Lumbar region is the predominant site of lesion, followed by the cervical region. Respected to Iacoangeli et al., this review articles with spinal secondarism of EW or general spinal intradural metastases were excluded, as having a homogenous summary.[31]
| Authors | Age | Sex | Location | EWS/FLI-1 | Surgery | CT | RT | PFS | OS | Outcome |
|---|---|---|---|---|---|---|---|---|---|---|
| Hisaoka et al.[34] | 14 | M | D12L2 | + | GTR | NA | NA | 3 m | 3 m | Alive |
| Isotalo[35] | 52 | M | L2L5 | NA | GTR | NA | CS | 12 m | 12 m | DF 1 m |
| Uesaka et al.[36] | 11 | F | C7D1 | NA | STR | NA | NA | NA | NA | NA |
| Akyüz et al.[37] | 31 | F | L1S2 | NA | STR | VCR, CCNU, CDDP | local | 2 m | 4 m | DOD 4 m |
| Mobley et al.[3] | 32 | M | L2L4 | + | STR | ActD+VDC/IE | local | 8 m | 12 m | DOD 12 m |
| Haresh et al.[4] | 26 | M | D11S2 | NA | GTR | VAC, ICE | Local | 2 m | 8 m | AWD 8 m |
| Kim and Shin[5] | 32 | F | C3C5 | NA | STR | IE | Local | 12 m | 12 m | DF 12 m |
| Klimo et al.[6] | 10 | M | L4S2 | NA | STR | VDC/IE | Local | 12 m | 12 m | DF 12 m |
| Yan et al.[7] | 10 | M | C2C3 | NA | GTR | - | - | 1 m | 1 m | DOD 1 m |
| Vincentelli et al.[8] | 40 | F | D11L4 | + | STR | DXR, holoxan | Local | 6 m | 6 m | DF 6 m |
| Duan et al.[9] | 8 | M | L2L4 | NA | + | NA | NA | NA | NA | Alive |
| Duan et al.[9] | 25 | M | L2L3 | NA | + | NA | NA | 6 m | 6 m | AWD 6 m |
| Karikari et al.[10] | 56 | F | L1 | + | GTR | VDC/IE | NA | NA | NA | DF |
| Pancucci et al.[11] | 55 | M | L4S2 | + | GTR | VIDE | Local | 13 m | 13 m | DF 13 m |
| Pancucci et al.[11] | 25 | F | L2L3 | + | GTR | - | - | 14 m | 14 m | Alive |
| Khalatbari et al.[12] | 28 | F | L5S1 | + | GTR | VDC/IE | local | 72 m | 72 m | DF 72 m |
| Bazzocchi et al.[13] | 44 | F | D6D7, L1L2 | NA | GTR | VDC/IE | Local | 31 m | 31 m | AWD 31 m |
| Huang et al.[14] | 39 | F | C4C6 | NA | GTR | VCR, THR, CTX | Local | 36 m | 36 m | Alive |
| Lozupone et al.[15] | 44 | F | L1S3 | NA | GTR | VDC/IE | Local | 6 m | 6 m | DF 6 m |
| Zhao et al.[16] | 14 | M | L2S1 | NA | STR | VDC | Local | NA | 12 m | DF 12 m |
| Bostelmann et al.[17] | 29 | M | C7 | + | GTR | VCR, IFO, DXR, VP16 | - | 1 m | NA | DF 18 m |
| Kartal and Akatlı[18] | 5 | M | D4D7 | NA | STR | NA | NA | NA | NA | NA |
| Chihak et al.[19] | 50 | M | D10L1 | + | GTR | VDC/IE | Local | 48 m | 60 m | DOD 60 m |
| Chihak et al.[19] | 60 | M | L2L3 | + | Partial | IE/AI | Local | 11 m | 48 m | DOD 48 m |
| Chihak et al.[19] | 25 | M | C4C7 | + | STR | VDC/IE | Local | NA | 20 m | DF 20 m |
| Chihak et al.[19] | 34 | M | L4L5, S1S2, S4S5 | + | Biopsy | VDC/IE | CS | 3 m | 3 m | DF 3 m |
| Scantland et al.[20] | 14 | F | L2L3 | + | STR | VDC/IE | Local | 24 m | 24 m | DF 24 m |
| Paterakis et al.[21] | 31 | M | L2L3, L5 | + | Partial | CPA, VCR, ADR, ActD, IFO, VP16 | - | 24 m | 42 m | Alive |
| Takami et al.[38] | 61 | M | L1L3 | + | GTR | VDC/IE | Local | NA | NA | Alive |
| Tan et al.[22] | 34 | F | C4T3 | + | Partial | CS | 9 m | 11 m | DOD 11 m | |
| Yan et al.[23] | 60 | M | D12L3 | - | GTR | + | - | NA | NA | NA |
| Izubuchi et al.[24] | 35 | F | D12L1, L4L5 | + | STR | VDC/IE | CS | 10 m | 16 m | DOD 16 m |
| Murray et al.[25] | 45 | M | L5S2 | + | GTR | + | Local | NA | NA | NA |
| Pu et al.[26] | 32 | M | D12L2 | + | GTR | VAC/IE | Local | 17 m | 17 m | DF 17 m |
| Ebrahimi et al.[27] | 13 | M | L1L2 | + | GTR | NA | NA | NA | NA | NA |
| Praveen et al.[28] | 23 | M | L1L2 | + | GTR | + | Local | 6 m | NA | NA |
| Shihadeh et al.[29] | 24 | M | C7D1 | + | STR | VAC/IE | Local | 6 m | 7 m | DOD 7 m |
| Salama et al.[30] | 58 | M | L3S1 | + | GTR | VAC/IE | Local | 13 m | 14 m | AWD 14 m |
| Current study | 36 | M | L2L5 | + | Partial | DXR+CPA | - |
NA: Not available, AWD: Alive with disease, DF: Disease free, DOD: Dead of disease, GTR: Gross total resection, STR: Subtotal resection, VCR: Vincristine, CDDP: Cisplatin, DXR: Doxorubicin, IFO: Ifosfamide, CPA: Cyclophosphamide, VP-16: Etoposide, Act-D: Actinomycin D, VDC: VCR+DXR+CPA, IE: IFO+VP-16, M: Male, F: Female, CS: Craniospinal, PFS: Progression-free survival (months), OS: Overall survival (months), IEES: Intradural extramedullary Ewing sarcoma, CT: Computed tomography, RT: Radiotherapy.
About 34% (n = 13) of the assessed 38 patients were female and 66% (n = 25) were male. The mean age of the cohort was 32.34 years, with patient ages ranging from 5 to 61 years. The prevailing site of disease was the lumbar spine (n = 27, 71%). The most common symptom mentioned by patients was pain. The mean OS of the cohort was 14.6 months.
As noted by Iacoangeli et al., available epidemiological data on IEES is likely unreliable, and there are currently no standardized clinical guidelines for its management in adults. Treatment approaches are often institution-specific.[31] However, the guidelines that may be most useful possible in the case of IEES are “Bone sarcomas: ESMO-PaedCanEURACAN” of 2021.[32] In a multidisciplinary collaboration between radiologists, pathologists, and surgeons working at a bone sarcoma reference center, new cases of bone tumors should be discussed. To evaluate the extent of the diseases, examinations such as bone scintigraphy and chest CT for general staging should be done. For staging, whole-body MRI or [18F] 2- fluoro-2-deoxy-D-glucose PET-CT or PETMRI are used more frequently.
The EE99 R3 study on primary disseminated multifocal ES identified several additional prognostic factors, including age at diagnosis (under 14 years), primary tumor volume >200 mL, the presence and number of bone lesions, additional pulmonary metastases, and bone marrow involvement. These factors were used to develop a more sensitive prognostic scoring system.[33] Renal, cardiac, and auditory dysfunction is a consequence/side effect of ChT. Therefore, before starting the treatment, it is essential to conduct baseline tests for renal and cardiac function and perform an audiogram (especially if platinum-based drugs are used). Standard ChT protocols typically include combinations of vincristine, doxorubicin, cyclophosphamide, ifosfamide, and etoposide.[20] Neoadjuvant ChT is strongly recommended, as moderate evidence supports its benefits in improving local control and long-term survival. Present trials consist of 3–6 cycles of initial combination ChT following biopsy, followed by local therapy, and an additional 6–10 cycles of ChT, typically administered at 2–3-week intervals. The total duration of the treatment is 10–12 months.
Local RT, with doses ranging from 30 to 54 Gy, may help prevent distant recurrence. Adjuvant RT is the preferred treatment for patients with non-sacral pelvic ES, regardless of surgical margins, tumor volume, or histological response. It has been shown to offer better local control and survival outcomes compared to surgery alone. Patients who received RT show a trend toward improved survival, although this effect is not statistically significant. RT may be particularly beneficial when adequate surgical margins cannot be achieved. However, caution is warranted with radiation therapy, as sarcomas secondary to RT after ES may develop in a dose-dependent manner. On the other hand, irradiating the lungs in patients with lung metastases may provide a survival advantage.
Primary IEES is more aggressive and carries a worse prognosis compared to its osseous counterpart,[23] appearing to be similar to conventional ES with metastases. Chihak et al. demonstrated that IEES has a median recurrence time of 18 months and a 58% 2-year event-free survival rate, despite multimodal therapy.[19] Other studies have reported a median progression-free survival of 12 months and OS of 14 months. Factors such as large tumor size, axial location, presence of metastases at the time of diagnosis, and positive surgical margins are all associated with poor overall survival. The influence of age on the prognosis of IEES is currently debated. These figures show that IEES has a worse prognosis compared to osseous ES.
Doxorubicin treatment is no longer recommended in relapsing ES, due to previously attained total doses. At this juncture, there is no standardized ChT treatment plan, and therefore substances such as alkylating agents (cyclophosphamide and high-dose ifosfamide) in combination with topoisomerase inhibitors (etoposide and topotecan), irinotecan with temozolomide or gemcitabine and docetaxel, or high-dose ifosfamide or carboplatin with etoposide are used.
Identifying local recurrence or metastases early during follow-up is important for increasing treatment success. For that reason, after completing ChT checks are suggested every 3 months for the first 2 years; every 6 months for 3–5 years; and every 6–12 months for 5–10 years.
As shown above, there is no standard protocol for IEES in the literature. However, we present a schematic flowchart that collects current management knowledge in IEES so as to be useful in daily clinical practice [Figure 3].
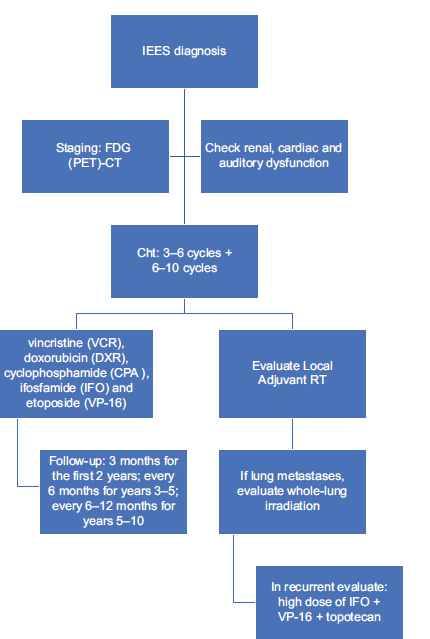
- Flowchart of intradural extramedullary Ewing sarcoma management. VCR: Vincristine, CDDP: Cisplatin, DXR: Doxorubicin, IFO: Ifosfamide, CPA: Cyclophosphamide, VP-16: Etoposide, FDG (PET): Fluorodeoxyglucose - positron emission tomography, RT: Radiotherapy, IEES: Intradural extramedullary Ewing sarcoma.
CONCLUSION
Despite advances in treatment modalities, the risk of local recurrence and distant metastasis underscores the need for radical surgery whenever achievable, close surveillance, and continued research to improve therapeutic strategies and outcomes for this rare entity. Primary IEES is an extremely rare condition with a similar presentation to spinal cord tumors. Available epidemiology is unreliable and therefore down to the present day, standard clinical guidelines for treatment are non-existent. Accordingly, multidisciplinary collaboration in specialty centers for bone sarcoma is pivotal for successful care. Neoadjuvant ChT as well as RT in addition to surgery are suggested. Follow-ups are crucial for early discovery of local-recurrences or metastases and contribute to improving long-term survival. Further studies are needed to portray primary IEES and postulate clinical guidelines for successful treatment.
Authors’ Contributions
AM, TDV: Study concept. TDV, GAI: Data acquisition. AM, FC: Quality control of data and algorithms. AM: Data analysis and interpretation. TDV, AMe: Manuscript preparation. AM: Manuscript editing. MML, LB, DG, FC: Manuscript review.
Ethical approval
The Institutional Review Board approval is not required.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship: Nil.
References
- The 2020 WHO classification of soft tissue tumours: News and perspectives. Pathologica. 2020;113:70-84.
- [CrossRef] [PubMed] [Google Scholar]
- Paravertebral “round cell” tumors in children. Radiology. 1969;92:1501-9.
- [CrossRef] [PubMed] [Google Scholar]
- Peripheral primitive neuroectodermal tumor/Ewing's sarcoma of the craniospinal vault: Case reports and review. Hum Pathol. 2006;37:845-53.
- [CrossRef] [PubMed] [Google Scholar]
- A rare case of intradural extramedullary Ewing's sarcoma with skip metastasis in the spine. Spinal Cord. 2008;46:582-4.
- [CrossRef] [PubMed] [Google Scholar]
- Primary intradural extraosseous ewing's sarcoma. J Korean Neurosurg Soc. 2009;45:179.
- [CrossRef] [PubMed] [Google Scholar]
- Primary pediatric intraspinal sarcomas. J Neurosurg Pediatr. 2009;4:222-9.
- [CrossRef] [PubMed] [Google Scholar]
- Intraspinal Ewing's sarcoma/primitive neuroectodermal tumors. J Clin Neurosci. 2011;18:601-6.
- [CrossRef] [PubMed] [Google Scholar]
- Primary intradural Ewing's sarcoma of the cauda equina presenting with acute bleeding. Acta Neurochir (Wien). 2010;152:563-4.
- [CrossRef] [PubMed] [Google Scholar]
- Intraspinal primitive neuroectodermal tumor: Imaging findings in six cases. Eur J Radiol. 2011;80:426-31.
- [CrossRef] [PubMed] [Google Scholar]
- Primary intradural extraosseous Ewing sarcoma of the spine: Case report and literature review. Neurosurgery. 2011;69:E995-9.
- [CrossRef] [PubMed] [Google Scholar]
- Primary extraosseous intradural spinal Ewing's sarcoma: Report of two cases. Acta Neurochir (Wien). 2013;155:1229-34.
- [CrossRef] [PubMed] [Google Scholar]
- Primary intradural extraosseous Ewing's sarcoma of the lumbar spine presenting with acute bleeding. Br J Neurosurg. 2013;27:840-1.
- [CrossRef] [PubMed] [Google Scholar]
- Intradural extramedullary Ewing's sarcoma recurrence with acute clinical presentation and literature review. Neuroradiol J. 2013;26:476-81.
- [CrossRef] [PubMed] [Google Scholar]
- Cervical primary ewing's sarcoma in intradural and extramedullary location and skip metastasis to cauda equine. Turk Neurosurg. 2015;25:943-7.
- [CrossRef] [Google Scholar]
- Magnetic resonance image findings of primary intradural Ewing sarcoma of the cauda equina: Case report and review of the literature. Spine J. 2014;14:e7-11.
- [CrossRef] [PubMed] [Google Scholar]
- Primary spinal intradural extraskeletal Ewing sarcoma mimicking a giant nerve sheath tumor: Case report and review of the literature. Int J Clin Exp Pathol. 2014;7:9081-5.
- [Google Scholar]
- The importance of surgery as part of multimodal therapy in rapid progressive primary extraosseous Ewing sarcoma of the cervical intra-and epidural space. Clin Pract. 2016;6:897.
- [CrossRef] [PubMed] [Google Scholar]
- Primary intradural extraosseous Ewing's sarcoma in a young child. Childs Nerv Syst. 2016;32:409-10.
- [CrossRef] [PubMed] [Google Scholar]
- Patterns of failure and optimal radiotherapy target volumes in primary intradural extramedullary Ewing sarcoma. Acta Oncol (Madr). 2016;55:1057-61.
- [CrossRef] [PubMed] [Google Scholar]
- Primary spinal intradural extraosseous Ewing sarcoma in a pediatric patient: Case report and review of the literature. Pediatr Neurosurg. 2018;53:222-8.
- [CrossRef] [PubMed] [Google Scholar]
- Intradural extramedullary Ewing's sarcoma: A case report and review of the literature. Neurol Neurochir Pol. 2017;51:106-10.
- [CrossRef] [PubMed] [Google Scholar]
- Primary intradural extramedullary Ewing sarcoma of the cervical spine: A case report and review of the literature. J Clin Neurosci. 2019;66:280-4.
- [CrossRef] [PubMed] [Google Scholar]
- Ewing's sarcoma in the spinal canal of T12-L3: A case report and review of the literature. Oncol Lett. 2019;18:6157-63.
- [CrossRef] [PubMed] [Google Scholar]
- Primary intradural extramedullary Ewing sarcoma: A case report and literature review. Oncol Lett. 2020;20:2347-55.
- [CrossRef] [PubMed] [Google Scholar]
- Primary intradural/extradural Ewing's sarcoma of the sacral spine: A case report and literature review. Surg Neurol Int. 2021;12:17.
- [CrossRef] [PubMed] [Google Scholar]
- Primary intradural extramedullary extraosseous Ewing's sarcoma/peripheral primitive neuroectodermal tumor (PIEES/PNET) of the thoracolumbar spine: A case report and literature review. Open Med. 2021;16:1591-6.
- [CrossRef] [PubMed] [Google Scholar]
- Primary intradural extramedullary Ewing sarcoma in the lumbar area: A case report. Radiol Case Rep. 2022;17:4617-21.
- [CrossRef] [PubMed] [Google Scholar]
- Primary spinal intradural extramedullary ewing's sarcoma/peripheral neuroectodermal tumour masquerading clinically as a neurogenic tumour: A case report and review of literature. Ann Neurosci. 2023;30:251-5.
- [CrossRef] [PubMed] [Google Scholar]
- Primary Ewing sarcoma of the cervical spine: A case report and literature review. Cureus. 2023;15:e42687.
- [CrossRef] [PubMed] [Google Scholar]
- Primary extraskeletal intradural Ewing sarcoma with acute hemorrhage: A case report and review of the literature. J Med Case Rep. 2024;18:144.
- [CrossRef] [PubMed] [Google Scholar]
- Nonmetastatic Ewing's sarcoma of the lumbar spine in an adult patient. Case Rep Oncol Med. 2012;2012:165289.
- [CrossRef] [PubMed] [Google Scholar]
- Bone sarcomas: ESMO-PaedCan-EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2018;29:iv79-95.
- [Google Scholar]
- Ewing sarcoma: Current management and future approaches through collaboration. J Clin Oncol. 2015;33:3036-46.
- [CrossRef] [PubMed] [Google Scholar]
- Peripheral primitive neuroectodermal tumour with ganglioneuroma-like areas arising in the cauda equina. Virchows Archiv. 1997;431:365-9.
- [CrossRef] [PubMed] [Google Scholar]
- Primary primitive neuroectodermal tumor of the cauda equina. Hum Pathol. 2000;31:999-1001.
- [CrossRef] [PubMed] [Google Scholar]
- Intradural, extramedullary spinal Ewing's sarcoma in childhood. J Clin Neurosci. 2003;10:122-5.
- [CrossRef] [PubMed] [Google Scholar]
- Primary primitive neuro-ectodermal tumor of cauda equina with intracranial seeding. Acta Neurochir (Wien). 2004;146:525-8.
- [CrossRef] [PubMed] [Google Scholar]
- Histologic features and prognosis of spinal intradural extramedullary Ewing sarcoma: Case report, literature review, and analysis of prognosis. World Neurosurg. 2018;115:448-52.e2.
- [CrossRef] [PubMed] [Google Scholar]




