
Translate this page into:
Uncommon dyselectrolytemia complicating Guillain–Barré syndrome
Address for correspondence: Dr. Aralikatte Onkarapa Saroja, Department of Neurology, KLE University's Jawaharlal Nehru Medical College and KLES’, Dr. Prabhakar Kore Hospital and MRC, Nehrunagar, Belgaum - 590 010, India. E-mail: sarojaao2002@yahoo.com
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Guillain–Barré syndrome (GBS) and hypokalemic paralysis are common causes of acute flaccid quadriparesis and specific therapeutic interventions differ. Simultaneous occurrence of severe hypokalemia in patients with GBS at the time of presentation can cause diagnostic and therapeutic dilemma. Presence of hypomagnesemia with hypokalemia in patients with GBS can be perplexing and pose further challenges. Evaluation for preexisting inherited or other associated metabolic disturbances is needed in the presence of such complex dyselectrolytemia. We report the rare association of GBS with severe hypokalemia and hypomagnesemia in a 41-year-old male presenting with acute flaccid quadriparesis and the therapeutic challenges faced.
Keywords
Guillain–Barré syndrome
hypokalemia
hypomagnesemia

Introduction
Hypokalemic paralysis and Guillain–Barré syndrome (GBS) are common differential diagnoses in patients presenting with acute flaccid paralysis.[1] Urgent recognition of hypokalemia is needed to prevent potentially lethal complications including cardiac arrhythmias and respiratory insufficiency.[2] Hypokalemia can be caused by acquired or inherited metabolic disorders of muscle ion channels and renal tubules.[2] Coexistence of hypokalemia, hypomagnesemia, and hypocalciuria occurs in Gitelman syndrome (GS), an inherited renal tubular disorder.[3] We document the coexistence of GBS with hypokalemia and hypomagnesemia akin to GS.
Case Report
A 41-year-old male presented with rapidly progressive symmetrical predominantly proximal quadriparesis of 4 days duration with tingling paresthesiae in limbs for 2 days. Productive cough for 2 days without fever occurred 1 week earlier. There was no history of alcohol consumption or exposure to drugs, bites, stings, and inoculations. He had no past history of similar symptoms, muscle aches, or thyroid dysfunction. There was no family history of neuromuscular diseases.
Heart rate was 88 beats/minute and blood pressure was 130/80 mmHg without respiratory insufficiency. He was awake, alert with symmetrical proximal muscle weakness (shoulders 2/5, elbow and lower limbs 4/5, wrist and fingers 5/5 by Medical Research Council [MRC] grading). There was mild weakness in the intrinsic hand muscles. Sensations, cranial and neck muscles were normal; muscle stretch reflexes sluggish and plantars flexor.
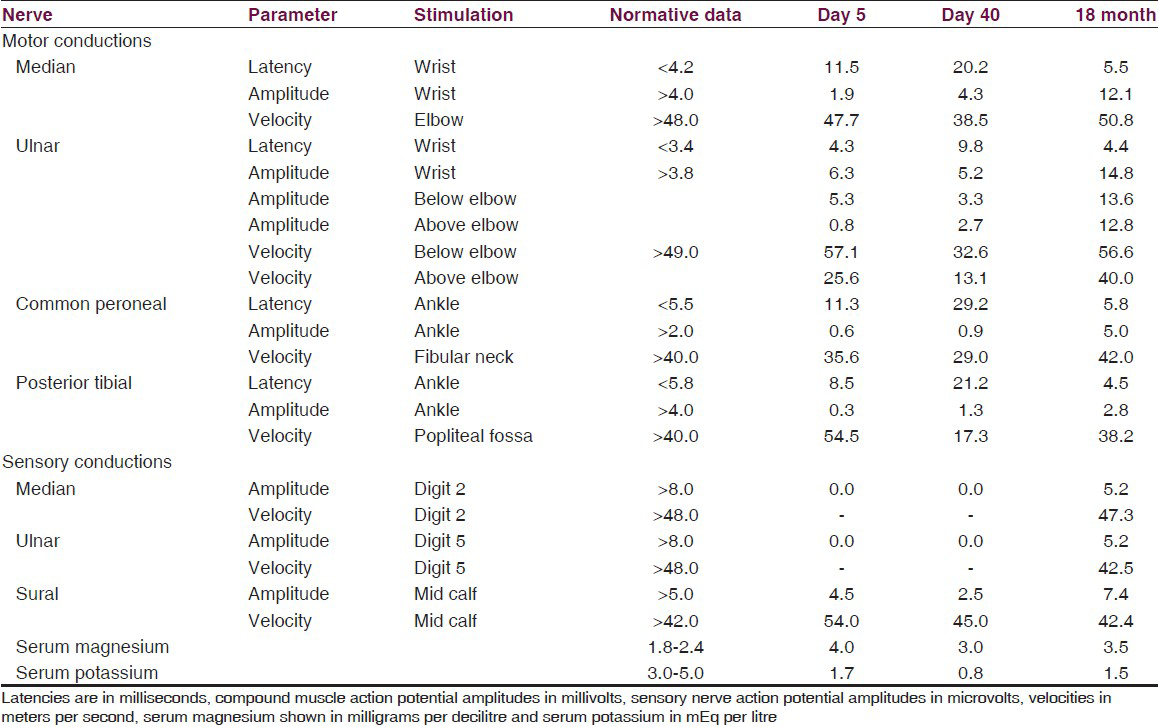
The serum potassium was 2.6 mEq/L, magnesium 1.7 mg/dL with mild metabolic alkalosis. Serum creatinine kinase (CK) was normal. Quadriparesis remained unchanged despite correction of hypokalemia to 4.0 mEq/L over 12 hours at which time nerve conduction studies (NCS) revealed increased distal motor latencies, partial conduction block along with nonlength-dependent reduction of amplitudes [Table 1]. Lumbar cerebrospinal fluid (CSF) revealed albuminocytological dissociation with 250 mg/dl protein and 20 cells/mm3 (lymphocytes 75%, polymorphs 25%).
Quadriparesis progressed with appearance of asymmetric lower motor neuron facial weakness and on fourth hospital day he required mechanical ventilation. Plasmapheresis was initiated on the third hospital day with 10 L removed over the next 9 days. Serum potassium and magnesium levels declined despite oral and parenteral supplements. Although patient could be weaned from ventilator after 12 days with moderate improvement of quadriparesis, he continued to have persistent hypotension and bradycardia requiring vasopressor support. The serum cortisol, thyroid hormones, CK-MB, echocardiography, and abdominopelvic imaging by ultrasound and contrast enhanced computerized tomography were normal.
Repeat nerve conductions before discharge revealed persistence of the abnormalities with increase in the latencies along with persistent dyselectrolytemia [Table 1]. At discharge 42 days after admission, the muscle power was 4/5 and he walked with minimal assistance. He was discharged on potassium supplements, spiranolactone, and dietary modification for hypomagnesemia. During follow-up the weakness completely recovered by 4 months. At the last follow-up 30 months after discharge, patient has persistent hypomagnesemia (1.5 mg/dl) with hypocalciuria (90 mg/day; normal 100-300 mg/day). Nerve conductions revealed significant improvement of distal latencies, amplitudes, and conduction velocities at 18 months [Table 1]. Serum magnesium and potassium levels in his siblings were normal.
Discussion
Acute neuromuscular paralysis is one of the common neurological emergencies. GBS and hypokalemia are commonly encountered causes of acute flaccid paralysis; other less common causes being hyperkalemia, botulism, porphyria, diphtheria, etc., Common subtypes of GBS consist of acute inflammatory demyelinating polyneuropathy (AIDP) and acute motor axonal neuropathy (AMAN) based on clinicopathological and electrophysiological findings. When the initial conduction studies are inconclusive, repeating the study after an interval would help in the differentiation between the subtypes.[1] Our patient had persistent hypokalemia and hypomagnesemia with clinical profile of AIDP supported by NCS findings and CSF showing albuminocytological dissociation.
Hypokalemia can result from excessive loss of potassium in the urine or from the gut, poor intake, increased translocation into cells or inherited tubular disorders.[4] Hereditary defects causing hypokalemia are Bartter's syndrome and GS.[24] The latter is an autosomal recessive renal tubular disorder due to mutations in the solute carrier family 12, member 3 gene SLC12A3, which encodes the thiazide sensitive sodium chloride cotransporter.[35] Diagnosis of GS is based on biochemical abnormalities characterized by hypokalemia, hypomagnesemia, hypocalciuria, and metabolic alkalosis. The prevalence of GS is estimated to be approximately 1:40,000 and is the most frequent inherited renal tubular disorder.[3] Though hypomagnesemia has been considered obligatory for the diagnosis of GS, few patients with severe hypokalemia without hypomagnesemia or hypocalciuria were proved to be suffering from GS by genetic studies.[23] Barter's syndrome is associated with hypokalemia and hypercalciuria[2] and presence of hypertension suggests hyperaldosteronism.[6]
Hypomagnesemia occurs in malnutrition, alcoholism, and with parenteral nutrition due to inadequate intake. Increased losses via kidneys, skin, gastrointestinal tract, or sequestration in the bone compartment also contribute to magnesium deficiency. Renal loss of magnesium could result from drugs.[2] Inherited renal magnesium wasting disorders include GS, Bartter's syndrome, isolated familial hypomagnesemia, hypomagnesemia with hypocalcaemia and hypomagnesemia with hypercalciuria. Neurological manifestations of hypomagnesemia include irritability, twitching, tremor and tetany. Cardiac dysfunction includes hypotension, arrhythmias, and sudden cardiac death. There is associated potassium and calcium abnormality confounding the cardiac and neurological manifestations.[27]
Our patient had severe hypokalemia with mild metabolic alkalosis when he was admitted with quadriparesis resulting from clinical, electrophysiological, and CSF features consistent with GBS. He had no predisposing acquired causes known to produce hypokalemia or hypomagnesemia. Reversible abnormalities in NCS with reduced amplitudes in motor and sensory conductions occur in patients with hypokalemic periodic paralysis.[89] During the course of the hospital stay, patient had recurrence of hypokalemia and hypomagnesemia that persisted during the follow-up despite the use of spiranolactone and potassium supplementation suggesting the presence of underlying metabolic dysfunction.
Plasmapheresis can cause transient hypomagnesemia[10] and possibly contributed to worsening of the already existing hypomagnesemia. However, presence of persistent hypomagnesemia and hypokalemia with hypocalciuria up to 30 months after completion of plasmapheresis suggests the presence of underlying abnormality of magnesium metabolism. The constellation of spontaneous hypomagnesemia, hypokalemia, hypocalciuria, and metabolic alkalosis is strongly suggestive of GS. Autonomic nervous system involvement in GBS can manifest with cardiac arrhythmia, blood pressure fluctuations, and is a relatively frequent occurrence.[1] These manifestations can also be caused by hypomagnesemia and hypokalemia.
Acute flaccid paralysis due to either hypokalemia or GBS manifesting as isolated entities may not perturb the clinician in diagnosis or management. Combination of both these conditions along with another comorbid dyseletrolytemia could be life-threatening. It is challenging to treat complex dyselectrolytemia like severe hypokalemia and hypomagnesemia, which can complicate the clinical burden of autonomic and neurological dysfunction in GBS. We report one such rare coexistence of complex dyselectrolytemia in a patient with GBS. While it is essential to prove GS by using genetic studies, the same could not be done due to lack of facility and remains a possible comorbid clinical entity.
Source of Support: Nil.
Conflict of Interest: None declared.
References
- Clinical disturbances of calcium, magnesium, and phosphate metabolism. In: Brenner BM, ed. Brenner and Rector's The Kidney (7th ed). Philadelphia: Saunders; 2004. p. :1041-76.
- [Google Scholar]
- Diagnosis of hypokalemia: A problem solving approach to clinical cases. Iran J Kidney Dis. 2008;2:115-22.
- [Google Scholar]
- Novel mutations in the thiazide-sensitive NaCl cotransporter gene in patients with Gitelman syndrome with predominant localization to the C-terminal domain. Kidney Int. 1998;54:720-30.
- [Google Scholar]
- Hypokalemic paralysis due to primary hyperaldosteronism simulating Gitelman's syndrome. Saudi J Kidney Dis Transpl. 2009;20:284-7.
- [Google Scholar]
- Gitelman's syndrome revisited: An evaluation of symptoms and health-related quality of life. Kidney Int. 2001;59:710-7.
- [Google Scholar]
- Dysfunction of sensory nerves during attacks of hypokalemic periodic paralysis. Neuromuscul Disord. 1999;9:227-31.
- [Google Scholar]
- Reversible electrophysiological abnormalities in hypokalemic periodic paralysis. Indian Pediatr. 2008;45:54-5.
- [Google Scholar]
- Profound ionised hypomagnesemia induced by therapeutic plasma exchange in liver cell failure patients. Transfusion. 2002;42:1598-602.
- [Google Scholar]




