
Translate this page into:
Suboccipital Extraspinal Extracranial Atypical Teratoid Rhabdoid Tumor
Address for correspondence: Dr. Ashok Gupta, Department of Neurosurgery, SMS Medical College, Jaipur - 302 004, Rajasthan, India. E-mail: drashokneuro@gmail.com
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Sir,
Atypical teratoid rhabdoid tumor (ATRT) is an uncommon malignant tumor predominantly affecting the pediatric population. The majority of the tumors are located within the cerebrospinal axis. Histopathologically, ATRT is characterized by rhabdoid tumor cells with cytoplasmic filamentous inclusions. The advent of molecular markers such as SMARCB1/integrase interactor 1 (INI1) has significantly improved our understanding of the tumor.
A 15-year-old man presented with complaints of neck pain and restriction of neck movement of 1-month duration which was insidious in onset and progressive in nature. There was also a significant loss of weight and appetite in the preceding 3 months. On examination, there was a swelling in the suboccipital region with restriction of neck movements, but the patient was neurologically intact. Contrast magnetic resonance imaging of the cervical spine showed a mass in the suboccipital region which was isointense on T1, hyperintense on T2, and intensely contrast enhancing [Figure 1]. There was no obvious intracranial or spinal extension or compression of the cervical spinal cord. Computed tomography cervical spine showed erosion of the squamous occipital bone, left occipital condyle, left half of arch of the atlas, and involvement of left atlanto-occipital joint. With a provisional diagnosis of soft tissue sarcoma or peripheral nerve sheath tumor, surgical resection of the tumor was planned. The tumor was yellowish gray, fish flesh-like, moderately vascular mass seen in the suboccipital region. Erosion of the squamous occipital bone left occipital condyle, left half of arch of the atlas, and involvement of left atlanto-occipital joint. Gross total excision was performed. The patient had an uneventful postoperative recovery and relief of the cervical pain. The histopathology and immunohistochemistry clinched the final diagnosis of ATRT. Interestingly, the tumor retained INI1 positivity [Figure 2]. The patient was then subjected to postoperative radiotherapy and continues to receive the same till the last follow-up.

- (a) Sagittal T2-weighted image showing hyperintense mass in the suboccipital region with no obvious intracranial or intraspinal extension. (b) Intense contrast enhancement on T1-weigted postcontrast image
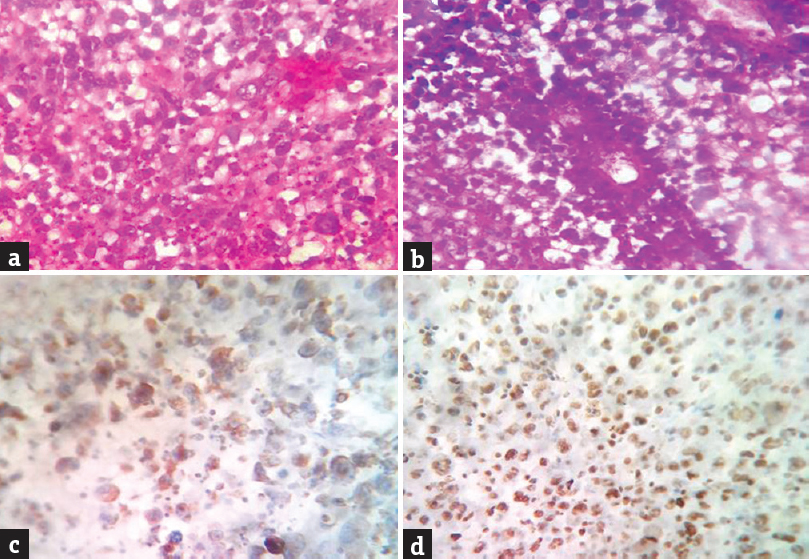
- (a) hematoxylin and eosin staining (×400) showing sheets of tumor cells. (b) hematoxylin and eosin staining (×400) perivascular pseudorosette formation. (c) immunohistochemistry (×400) vimentin positivity. (d) immunohistochemistry (×400) integrase interactor 1 positivity
ATRT was first described by Roker in 1996. It was included by the WHO in brain tumor classification later in the year 2000. They are rare, aggressive, and highly malignant embryonal tumors of the nervous system.[1] They are most prevalent in the pediatric age group under the age of three accounting for approximately 1%–2% of pediatric brain tumors. Only 26 cases have been reported in adults (18 years of age or older)[2] ATRT is associated with an extremely poor prognosis with a mean overall survival ranging from 6 to 18 months.[1] Intracranial ATRTs are commonly located in the cerebral hemispheres, while infratentorial neoplasms are seen in the cerebellum and are relatively rare. It is uncommon for ATRT to be located in the spinal cord of adults or children.[2] They have been reported in a myriad of sites within the craniospinal axis (suprasellar region, along with cranial nerves II, III, and VII, in cerebellopontine angle, cisterns, and pineal region). To the best of our knowledge, ATRT in the suboccipital and cervical region has not been reported in literature. Histopathologically, ATRT is characterized by rhabdoid tumor cells, which have vesicular nuclei, large nucleoli, and cytoplasmic filamentous inclusions. Besides the presence of rhabdoid cells, the lack of nuclear INI1 expression is the most important prerequisite to secure the diagnosis and is usually used as an important marker to distinguish from other central nervous system tumors. The majority of ATRT cases display genetic alterations of SMARCB1 (INI1/hSNF5), a tumor suppressor gene located on 22q11.2, and resulting in loss of INI1 protein. INI1 protein is ubiquitously expressed in the nuclei of all normal cells and can be identified using immunohistochemistry. The subsequent loss of INI1 protein expression comprises a relatively specific and sensitive diagnostic marker for ATRT.[3] ATRT to retain the SMARCB1/INI1 positivity is a very atypical and rare phenomenon. Bertozzi et al. have reported two cases of ATRT in which SMARCB1/INI1 staining was retained. One of these patients had loss of expression of another SWI/SNF chromatin-remodeling complex member, the ATPase subunit SMARCA4/BRG1 due to a homozygous mutation.[4] Our patient also had retained the INI1 positivity, but the genetic analysis could not be performed. Over the past few decades, despite advances in therapeutic approaches, the outcome remains dismal. Radiotherapy till date seems to provide the best possible chance to salvage these patients. As it predominantly affects the pediatric age group, the actual role of radiotherapy becomes very limited. Survival seems to be better with focal radiation with intrathecal chemotherapy being useful in patients who are not candidates for radiation. There is no accepted standard chemotherapeutic regimen, but intensive alkylator-based regimens with stem cell rescue may be effective in these patients. Repeat surgery and radiation are required more often than not in patients receiving chemotherapy. As our understanding of the molecular mechanisms of the disease continues, molecular targets are being explored as therapeutic options.[5] ATRTs are very aggressive tumors with a dismal outcome despite aggressive multimodality treatment. The possibility of ATRT should be considered in the differential diagnosis of tumors in unusual locations. Loss of INI1 should not be solely relied upon for the diagnosis as few cases may retain the INI1 positivity on immunohistochemistry as highlighted in the current case. These extracranial, extraspinal cases of ATRT with INI1 positivity may be a separate subtype of the ATRT. Further research at the molecular and genetic level may provide more knowledge about this variant.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- Atypical teratoid rhabdoid tumors: A population-based clinical outcomes study involving 174 patients from the surveillance, epidemiology, and end results database (1973-2010) Cancer Manag Res. 2015;7:301-9.
- [Google Scholar]
- Atypical teratoid/rhabdoid tumor of the central nervous system in an 18-year-old patient. Clin Neuropathol. 2009;28:1-10.
- [Google Scholar]
- Atypical teratoid rhabdoid brain tumor in an infant with ring chromosome 22. Korean J Pediatr. 2014;57:333-6.
- [Google Scholar]
- Atypical teratoid rhabdoid tumours (ATRT): A French retrospective study. Neuro Oncol. 2014;16:i1-9.
- [Google Scholar]
- Atypical teratoid rhabdoid tumor: Current therapy and future directions. Front Oncol. 2012;2:114.
- [Google Scholar]




