
Translate this page into:
Pleomorphic xanthoastrocytoma in a case of tuberous sclerosis
Address for correspondence: Dr. Mutum Samarendra Singh, Department of Pathology, Hospital Universiti Sains, Kubang Kerian, 16150, Kelantan, Malaysia. E-mail: mutum@kb.usm.my
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Tuberous sclerosis is a known phakomatosis and the associated finding of a subependymal giant cell astrocytoma is common with this disorder. A case of tuberous sclerosis with a finding not previously reported, i.e. that of a pleomorphic xanthoastrocytoma, is presented here.
Keywords
Cystic tumor
pleomorphic xanthoastrocytoma
tuberous sclerosis

Introduction
Tuberous sclerosis is a known entity being commonly described along with the other phakomatoses with established associations with a particular type of astrocytoma, typically the SEGA variety (subependymal giant cell astrocytoma). Here we present a case of tuberous sclerosis with a finding not previously reported, i.e. that of a pleomorphic xanthoastrocytoma.
Case Report
A 23-year-old male who was diagnosed with tuberous sclerosis at an early age came to our attention following complaints of blurring of vision for 1 month, associated with nausea, vomiting and headaches. On examination, he was found to be alert with full (GCS) score and bilateral papilledema. His vision was compromised with perception of hand movements noted in both eyes and Shagreen patches were noted over his back.
A CT brain scan was done which revealed a right supratentorial intra-axial lesion which was largely cystic with an enhancing mural nodule as depicted in Figure 1. Additionally, midline shift with mild perilesional vasogenic edema were also noted. From the imaging findings, differential diagnoses in this case at this point were subependymal giant cell astrocytoma and pilocytic astrocytoma.
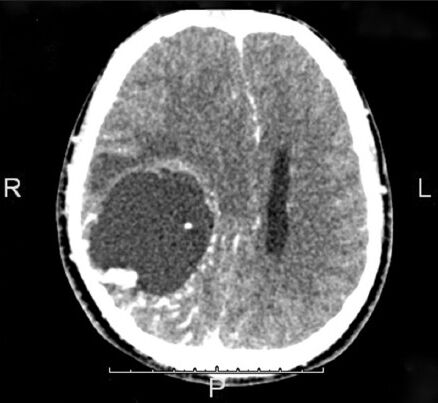
- The CT image showing the presence of the intra-axial space occupying lesion in the right hemisphere
A craniotomy and a total excision of tumor was performed which was found to be cystic and with minimal solid components. Post-operatively the patient remained alert and symptom free apart from visual compromise. The tumor samples were sent for histopathological examination and were found to be pleomorphic xanthoastrocytoma.
As seen in the Figure 2a–d, sections from the wall of the cystic mass showed mildly pleomorphic cells having round to ovoid nuclei with occasional lobulated outline and eosinophilic cytoplasm in a fibrillary background and absent mitotic figures. Scattered lymphocytes and foamy macrophages were noted and hyaline globules and Rosenthal fibers were not seen in this specimen. The tumor cells showed strong positivity for GFAP) and S100 as well.
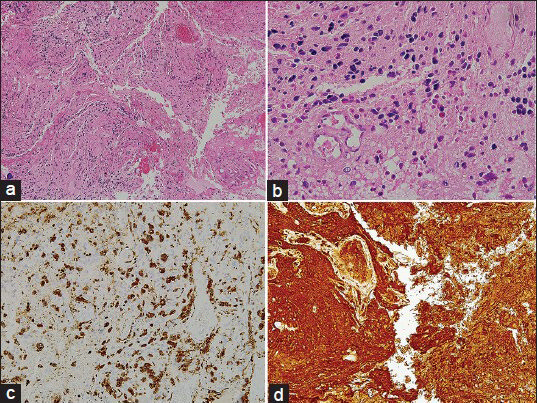
- The CT image shown above notes the presence of the intra-axial space occupying lesion in the right hemisphere. Above Right (a): H and E, ×100,Above Left (b): H and E, ×400, Below Right (c): CD 68 staining depicting positivity for macrophages, Below Left (d): GFAP staining depicting positivity
Discussion
Tuberous sclerosis is a known entity being commonly described along with the other phakomatoses and has established associations with a particular type of astrocytoma, typically the SEGA variety. Tuberous sclerosis aka Bournville Disease is a group of autosomal dominant disorders characterized by hamartomas and benign neoplastic lesions that affect the central nervous system and various non-neural tissues. This disorder affects around 25 000 to 40 000 individuals in the United States and about 1-2 million individuals worldwide.[1] Apart from known manifestations seen in the skin, eyes, kidney, heart, digestive system and lung, the central nervous system manifestations of tuberous sclerosis are cortical tubers (freq 90-100%), subependymal nodules (90-100%) and white matter hamartomas and heterotopias (90-100%). The variety of astrocytomas commonly associated with tuberous sclerosis is the SEGA with incidences ranging from 6% to 16%. This findings in this case is unique in that the variant of astrocytoma is hitherto not reported nor described in the current literature, i.e. that of a pleomorphic xanthoastrocytoma.
Pleomorphic xanthoastrocytoma is a rare primary neoplasm of the central nervous system, accounting for only 1% of all astrocytic tumors and was originally described by Kepes et al. in 1979 in his study of 12 cases.[2] This tumor tends to be typically superficially located, supratentorial with a predilection for involving the temporal lobes. It is characteristically seen in young adults who present with intractable seizures, and rendered seizure free post GTR (gross total resection or within 1 year of surgery.[34] Pleomorphic xanthoastrocytomas typically appear as hypointense cystic masses with an enhancing mural nodule on T1-weighted MRI, hyperintense or mixed signal intensity on T2 weighted MRI with suppression of cyst content on FLAIR images.[5] Additionally, Raz et al. also described the peculiar finding of enhancement of the adjacent meninges, which is seen in up to 70% of cases.[5] This tumor is of a low grade subtype, classified under WHO Grade 2 group of gliomas, and although malignant transformation into a higher grade or anaplastic variant is considered rare, histologically this tumor can be confused with Grade IV giant cell glioblastoma due to the findings of giant cells. Pleomorphic xanthoastrocytomas are somewhat uncommon and typically slow-growing, and cases which are completely excised show favorable prognosis.
Classic histopathologic features included an admixture of mononuclear cells, bizarre multinucleated giant cells, spindled cells and lipid-rich vacuolated cells. The tumor cells were associated with abundant lymphocytes and reticulin fibers. They showed little tumor necrosis or mitotic activity. Immunohistochemical study demonstrated diffuse positive staining for glial fibrillary acidic protein, vimentin and S-100 protein.[167] Seventy-seven percent of the cases also showed positive staining for CD34 and it has been suggested that CD34 staining may be used to guide differential diagnoses of these lesions.[8] Mitotic activity has been reported to be uncommon in pleomorphic xanthoastrocytomas and some investigators have highlighted its prognostic significance, as it was the most significant predictor of recurrence and survival.[9] Although surgery, specifically gross total resection, is the treatment of choice, wherein 10 year survival of 70-80% can be expected, currently there is no consensus regarding the role of adjuvant therapy or radiotherapy.[910] It is however interesting to note that the single most significant predictor of recurrence free survival is the extent of resection as reported by Gianni et al.[9] Recurrence free survival was significantly longer in cases who have undergone gross total resection (>80% at 15 years) as compared to subtotal resection (50% at 15 years).[7]
Powell et al. explored the possibility of divergent differentiation in pleomorphic xanthoastroytomas, noting a possible relationship to ganglion cell tumors.[10] The expression of glial and neuronal polypeptides was examined in routinely prepared surgical resections by immunohistochemistry using well-characterized antibodies that recognize GFAP, synaptophysin (SYN), and neurofilament triplet polypeptides (NFPs) in microwave-enhanced single-and double-immunolabeling experiments. Each neoplasm contained cells that were immunoreactive for SYN and/or NFPs, GFAP, and occasionally for both GFAP and either NFP or SYN, and concluded that abortive neuronal/neuroendocrine differentiation may occur in PXAs, suggesting a relationship between PXA and other developmental neoplasms that reveal a more overt neuronal phenotype, such as ganglioglioma, dysembryoplastic neuroepithelial tumor, and desmoplastic ganglioglioma, and with tumors expressing ambiguous glial/neuronal lineage, such as the subependymal giant cell tumor of tuberous sclerosis. Powell et al. also suggested that aberrant expression and accumulation of neuronal intermediate filaments may account for the large, pleomorphic cell morphology observed in many of these tumors.[10]
Source of Support: Nil.
Conflict of Interest: None declared.
References
- WHO Diseases of the central nervous system. Lyon: International Agency for Research on Cancer (IARC); 2007.
- [Google Scholar]
- Pleomorphic xanthoastrocytoma: A distinctive meningocereberal glioma of young subjects with relatively favorable prognosis. A study of 12 cases. Cancer. 1979;44:1839-52.
- [Google Scholar]
- Pleomorphic xanthoastrocytoma, a distinctive astroglial tumor: Neuroradiologic and pathologic features. AJNR Am J Neuroradiol. 1993;14:1397-404.
- [Google Scholar]
- MRI of temporal lobe pleomorphic xanthoastrocytoma. J Comput Assist Tomogr. 1992;16:856-9.
- [Google Scholar]
- Pleomorphic Xanthoastrocytomas: What do we really know About It? Cancer. 1999;85:2033-45.
- [Google Scholar]
- Xanthoastrocytoma: Favourable outcome after complete surgical resection. Neuro Oncol. 2001;3:184-92.
- [Google Scholar]
- Pleomorphic Xanthoastrocytoma: Report on four cases-clinical, radiologic and pathologic features. Neurosurg Rev. 1991;14:43-9.
- [Google Scholar]
- Pleomorphic xanthoastrocytoma and its Differential Diagnosis: A Study of Five Cases. Hum Pathol. 1991;22:1128-35.
- [Google Scholar]
- Differentiation in pleomorphic xanthoastrocytomas: Evidence for a neuronal element and the possible relationship to ganglion cell tumors. Am J Surg Pathol. 1996;20:80-5.
- [Google Scholar]




