
Translate this page into:
Pineal region pilocytic astrocytoma; an unusual site with variable radiological, clinical, and histological features: A report of two cases
Address for correspondence: Dr. Vineeta Vijay Batra, Department of Pathology, G B Pant Institute of Postgraduate Medical Education and Research, Jawahar Lal Nehru Marg, New Delhi - 110 002, India. E-mail: vvbatra9@gmail.com
This is an open access article distributed under the terms of the Creative Commons Attribution NonCommercial ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non commercially, as long as the author is credited and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Sir,
Pineal region tumors account for <1% of all intracranial neoplasms, of which approximately 14–27% is of pineal parenchymal origin.[1] Gliomas are very rare in the pineal region. They are thought to arise from the native glial cells of pineal gland or from the adjacent structure.[2] A wide spectrum of pineal region glial tumors have been described including pilocytic astrocytoma (PA), pleomorphic xanthoastrocytoma, glioblastoma, oligodendroglioma, and ganglioglioma; mainly as case reports. We report two cases of pineal region PA with variable clinical, radiological, and histological features with a review of literature.
Case 1: An 11-year-old female child was brought to the Neurosurgery Outpatient Department with the chief complaints of headache, retro-orbital pain, vomiting, and bilateral diminution of vision for 1 month. Contrast-enhanced magnetic resonance imaging with magnetic resonance spectroscopy revealed a mass in the pineal region measuring 40 mm × 33 mm × 29 mm (arrow), showing iso- to hypo-intense signals on T1-weighted and hyperintense signals on T2-weighted images with marked peripheral enhancement (arrow) on postcontrast scan [Figure 1]. The spectroscopy from the peripheral part of the lesion shows elevated choline peak (arrow), favoring a malignant lesion, likely pineoblastoma.
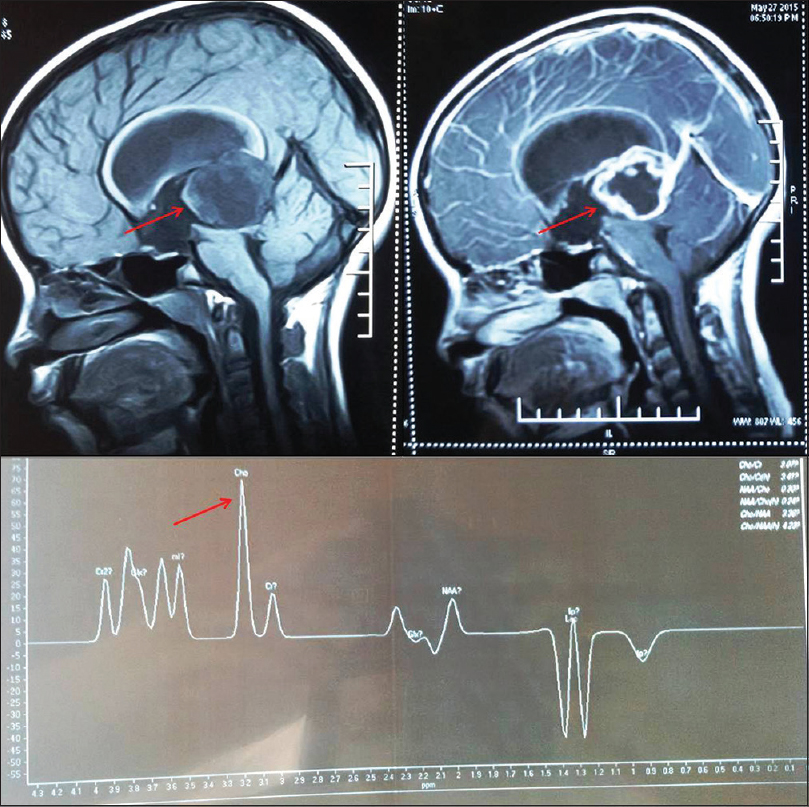
- Contrast-enhanced magnetic resonance imaging with magnetic resonance spectroscopy revealed a mass in the pineal region showing iso- to hypo-intense signals on T1-weighted (arrow) and hyperintense signals on T2-weighted images with marked peripheral enhancement (arrow) on postcontrast scan. The spectroscopy from the peripheral part of the lesion shows elevated choline peak (arrow)
Near total excision of the mass was performed through sub-occipital craniotomy and supracerebellar infratentorial approach. The tumor was highly vascular and soft, with well-defined plane of cleavage. An intraoperative frozen section showed a glial tumor comprising bipolar piloid cells admixed with numerous eosinophilic granular bodies (EGBs) and Rosenthal fibers [Figure 2a] characteristic of PA.
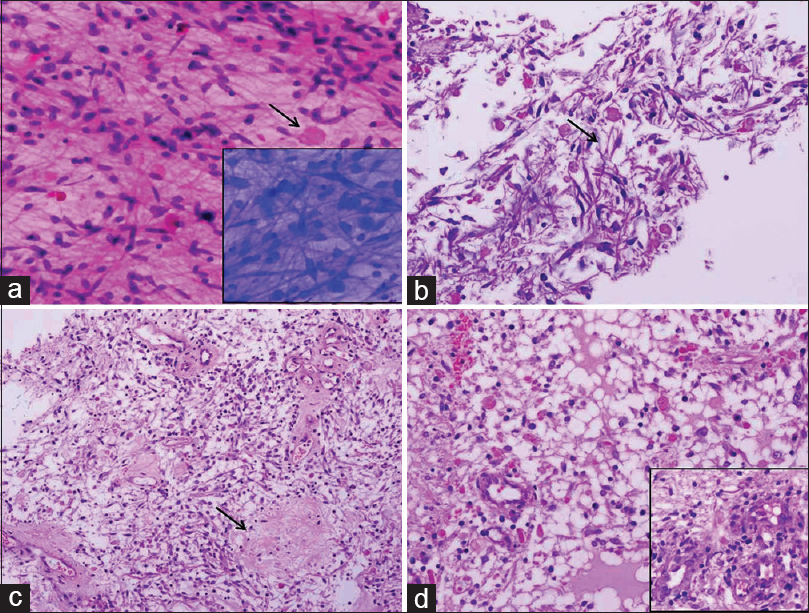
- Photomicrographs showing histomorphologic features of Case 1. (a) Frozen section showing a glial tumor comprising bipolar piloid cells admixed with numerous eosinophilic granular bodies (arrow), inset showing toluidine blue image (H and E, ×200), (b) paraffin section of the frozen tissue showing piloid cells in a fibrillary background with abundant Rosenthal fibers (arrow) and eosinophilic granular bodies (H and E, ×200), (c and d) main tissue sections displaying hypocellular (arrow) and hypercellular areas with glomeruloid vessels in the inset (H and E, ×200)
Paraffin sections showed a glial tumor displaying both hypocellular and hypercellular areas comprising mainly of piloid cells with extensive Rosenthal fibers and EGBs [Figure 2b–d]. Few glomeruloid vessels were also noted. No pleomorphism, mitotic activity, or necrosis was seen. A final diagnosis of PA WHO Grade I was made. The patient was discharged with improved vision and is doing well at a 2-month follow-up.
Case 2: A 22-year-old young female was admitted with the chief complaints of gradually progressive severe headache for 6 months and altered sensorium and recurrent episodes of seizures for the past 1 week. On examination, subnormal higher mental function, altered behavior, difficulty in walking, and decreased hearing in bilateral ears were noted.
The contrast-enhanced computer tomography scan showed a large well-defined predominantly cystic mass with peripheral coarse calcification in the pineal region measuring 8.7 cm × 8 cm × 7.8 cm [Figure 3]. The mass extended into the 3rd ventricle and lateral ventricles with effacement of the 4th ventricle. A radiological diagnosis of pineoblastoma was offered. The tumor was cystic and filled with jelly-like material. Subtotal resection of the cyst was done.
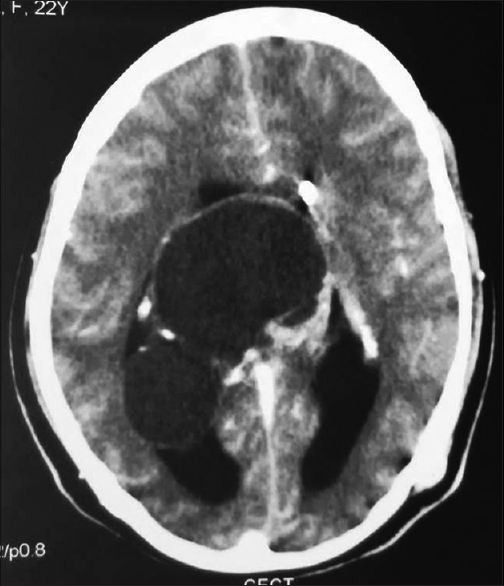
- Contrast-enhanced computer tomography scan showing a large well-defined cystic mass with peripheral coarse calcification in the pineal region extending through the 3rd ventricle to involve both lateral ventricles and compressing medial part of the bilateral cerebellar hemispheres with the effacement of the 4th ventricle
Light microscopy showed a glial tumor comprising both cellular and hypocellular areas. The hypocellular areas showed piloid cells in a fibrillary background with areas of calcification, glomeruloid vascular proliferation, and occasional EGBs [Figure 4a and b]. The cellular areas delineated an oligodendroglioma-like morphology [Figure 4c]. Focal areas of foam cell change were also noted. No mitosis or necrosis was seen. Fluorescent in situ hybridization showed normal 1p and 19q signal pattern ruling out the possibility of oligodendroglioma. The immunohistochemistry for glial fibrillary acidic protein (GFAP) showed diffuse fibrillary positivity [Figure 4d], whereas synaptophysin, p53, and neurofilament protein were negative. MIB-1 labeling index was 1–2%. A final diagnosis of PA WHO Grade I was made. The patient was symptom-free at 1-month follow-up.
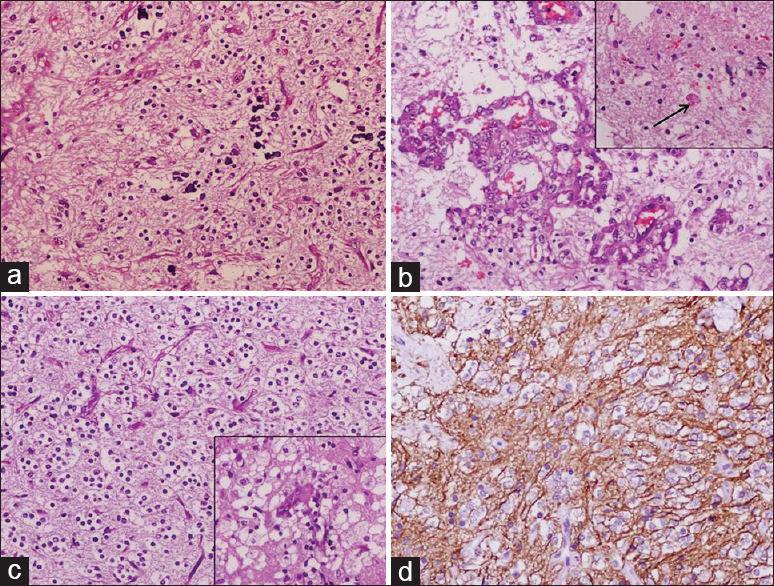
- Photomicrographs showing histomorphology and immunohistochemistry features of Case 2. (a) The hypocellular area of the tumor showing piloid cells in a prominent fibrillary background with areas of calcification (H and E, ×200), (b) tumor showing glomeruloid vascular proliferation and occasional eosinophilic granular body in the inset (arrow), (c) the cellular area delineated an oligodendroglial-like morphology and focal area of foam cell change in the inset (H and E, ×200), (d) glial fibrillary acidic protein stain showing diffuse fibrillary positivity (×200)
It is a matter of debate whether glial tumors of the pineal gland arise primarily from it or involve it secondarily.[3] Papasozomenos first described the presence of GFAP-positive cells in the human pineal gland.[2] Most cases of pineal glial tumors reported in literature showed extension to adjacent structures and ventricles. The tumor in both our cases also showed extension into ventricles. Praver et al. suggested that in addition to the native astrocytes and interstitial cells of the pineal gland, ependymal cells of ventricles and glial cells from the brainstem may also contribute to tumor mass.[4] It is possible that the range of tumor histology encountered among these pleomorphic neoplasms is a result of varying degrees of contribution from local cell populations.
Pineal region tumors encompass a wide spectrum of histology ranging from benign to malignant. Case 1 was a solid tumor with a characteristic histomorphology of PA, whereas Case 2 was predominantly a cystic mass with widespread involvement of adjacent structures and on histology showed oligodendroglioma-like areas. An accurate intraoperative frozen section diagnosis accompanied with a confirmatory paraffin section diagnosis is essential for optimal clinical management. However, sometimes, it can be difficult to offer accurate diagnosis more so in frozen section because of the propensity for mixed tumor pathology and heterogeneity in the pineal region.
Gliomas of the pineal region are reported to be of two varieties, distinguishable by the presence or absence of a cyst. The cystic variant is reported to have a more benign clinical course and tends to be restricted in the pineal gland. However, in our Case 2, though tumor was largely cystic, it showed a widespread extension into the adjacent structures.
Most of these patients present with symptoms of increased intracranial pressure. Patients may also present with Parinaud's syndrome and less commonly, gait disturbances, seizures, and cranial nerve palsies. The favorable prognosis of these tumors in the absence of characteristic radiological appearance emphasizes the role for histological confirmation of all pineal region tumors. The epiphysis has a propensity to form Rosenthal fibers.[5] Hence, low-grade glial tumors should be carefully and appropriately distinguished from nonneoplastic fibrillary gliosis of the pineal region.
Pineal cysts are a common imaging finding with a reported frequency of 25–40% in autopsy series.[6] It is difficult to distinguish low-grade cystic gliomas in the pineal region from the more common pineal cyst. However, due to overlapping radiological and clinical presentations, histological examination is important to differentiate between the two.
To conclude, given the diverse clinical presentations, overlapping radiological features, and differences in their management and outcomes of the tumors encountered in the pineal region, an accurate intraoperative frozen section and postoperative pathological diagnosis are mandatory.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest
Acknowledgment
We would like to acknowledge Dr. Deepti for helping in data collection.
References
- WHO classification of tumours of the central nervous system. In: Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, eds. Pineocytoma (4th ed). Lyon: IARC Press; 2007. p. :122.
- [Google Scholar]
- Glial fibrillary acidic (GFA) protein-containing cells in the human pineal gland. J Neuropathol Exp Neurol. 1983;42:391-408.
- [Google Scholar]
- Pineal gland lesions: A cytopathologic study of 20 specimens. Cancer. 2005;105:80-6.
- [Google Scholar]
- Atypical pleomorphic neoplasms of the pineal gland: Case report and review of the literature. Surg Neurol Int. 2015;6:129.
- [Google Scholar]
- Pineal astrocytoma: Report of a case, confined to the epiphysis, with immunocytochemical and electron microscopic studies. Cancer. 1981;47:99-103.
- [Google Scholar]
- High prevalence of pineal cysts in healthy adults demonstrated by high-resolution, noncontrast brain MR imaging. AJNR Am J Neuroradiol. 2007;28:1706-9.
- [Google Scholar]




