
Translate this page into:
Myelin oligodendrocyte glycoprotein associated disease presenting as rhombencephalitis with horizontal gaze palsy
*Corresponding author: Rajarshi Chakraborty, King George Medical University, Lucknow, Uttar Pradesh, India. satyalung@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Verma R, Bal KP, Chakraborty R. Myelin oligodendrocyte glycoprotein associated disease presenting as rhombencephalitis with horizontal gaze palsy. J Neurosci Rural Pract. 2025;16:115-7. doi: 10.25259/JNRP_268_2024
Abstract
Myelin oligodendrocyte glycoprotein associated disease (MOGAD) has a variety of manifestations spanning from optic neuritis to acute disseminated encephalomyelitis-like presentations. Rhombencephalitic presentation is an unusual entity in MOGAD. A 36-year-old male presented with sub-acute onset diplopia, and facial and bulbar palsy with alteration in sensorium following febrile illness. The workup revealed demyelinating lesions, subtle optic neuritis, and positive antimyelin oligodendrocyte glycoprotein antibodies. The patient showed significant improvement with pulse steroid therapy. This case illustrates the prudentiality of early diagnosis and treatment of MOGAD which can give positive outcomes.
Keywords
Horizontal gaze palsy
Myelin oligodendrocyte glycoprotein associated disease
Outcome
Rhombencephalitis

INTRODUCTION
Myelin oligodendrocyte glycoprotein associated disease (MOGAD) is a relatively newer entity with a plethora of manifestations including optic neuritis (most common), transverse myelitis, acute disseminated encephalomyelitis (ADEM), ADEM-like presentations, FLAMES (fluid-attenuated inversion recovery [FLAIR]-hyperintense lesions in myelin oligodendrocyte glycoprotein [anti-MOG] associated encephalitis with seizures) and other demyelination syndromes.[1,2] MOGAD presenting as rhombencephalitis is relatively uncommon. Henceforth, we discuss such a presentation in MOGAD.
CASE REPORT
A 36-year-old male presented with a fever for three days followed by diplopia, right-sided-facial palsy, along with dysphagia, and dysarthria with nasal intonation. He also developed an alteration of sensorium on the fifth day. However, there was no history of seizures, emotional lability, forgetfulness, headache, and difficulty in micturition/defecation. Past and family histories were insignificant.
General examination was unremarkable with normal vitals. His nervous system examination revealed a Glasgow coma score of 12 (E3V4M5), with hypo-responsiveness (drowsy). Cranial nerve examination showed complete horizontal ophthalmoplegia with preserved vertical eye movements with normal fundus examination, right-sided lower motor neuron type of facial palsy [Figure 1a and Supplementary video 1]. The patient also had reduced sensation over the right half of the face with reduced movements of the right side of the palate. Motor examination showed right-sided hemiparesis with the power of grade 4 on the right side. Heel–shin test was abnormal in the right lower limb, the normal finger nose test, and the tandem gait was abnormal. The rest of the nervous system examination and other system examinations were unremarkable. Visual evoked potentials had prolonged P-100 latencies in both eyes (114 milliseconds in the right eye and 126 milliseconds in the left eye) [Figure 1b].
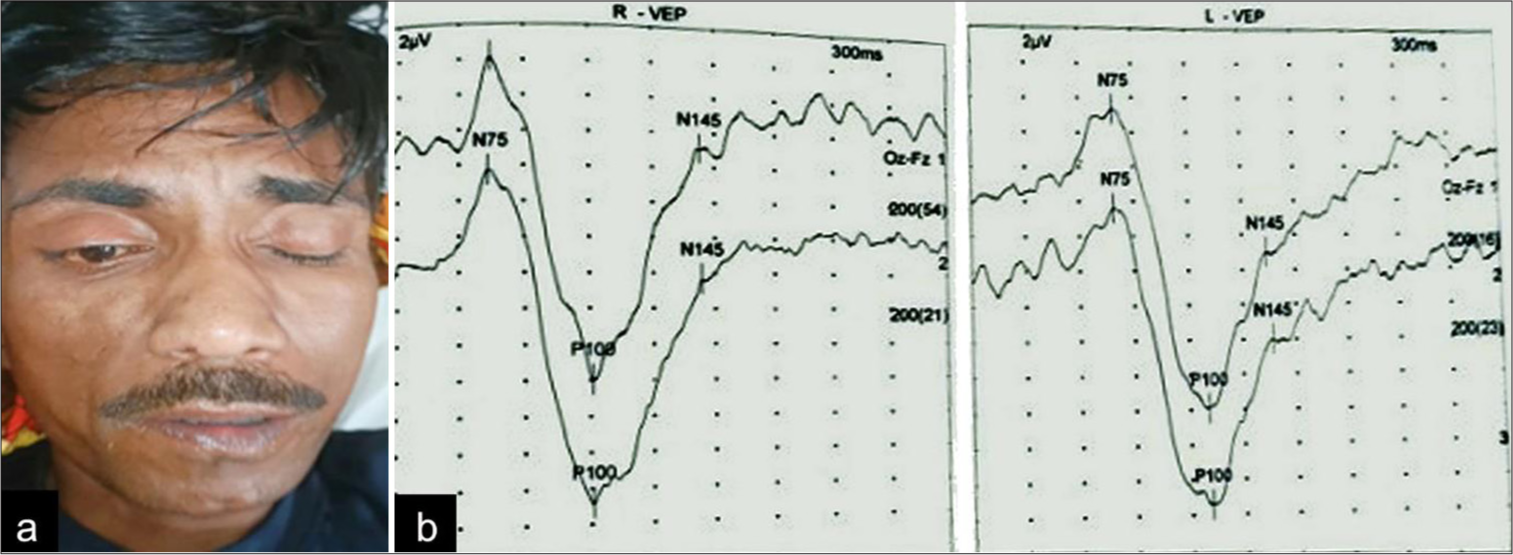
- (a) Incomplete closure of right eye with flattening of the right nasolabial fold; (b): Visual evoked potentials (VEP) showing prolonged P-100 latencies in both eyes (114 milliseconds in the right eye and 126 milliseconds in the left eye).
Complete hemogram, electrolytes, liver, renal, and thyroid function tests were within normal limits. In a cerebrospinal fluid study, total cells were <5 cells/mm3 with normal protein and glucose levels. Magnetic resonance imaging of the cranium revealed T2-FLAIR sequence non-enhancing fluffy hyperintensities involving mainly pontine tegmentum [Figure 2]. Anti-MOG antibody was positive and the anti-aquaporin antibody was negative. The diagnostic workup of rhombencephalitis was done including septic, inflammatory, demyelination, vascular, neoplastic, and metabolic causes. Genetic testing was not performed.
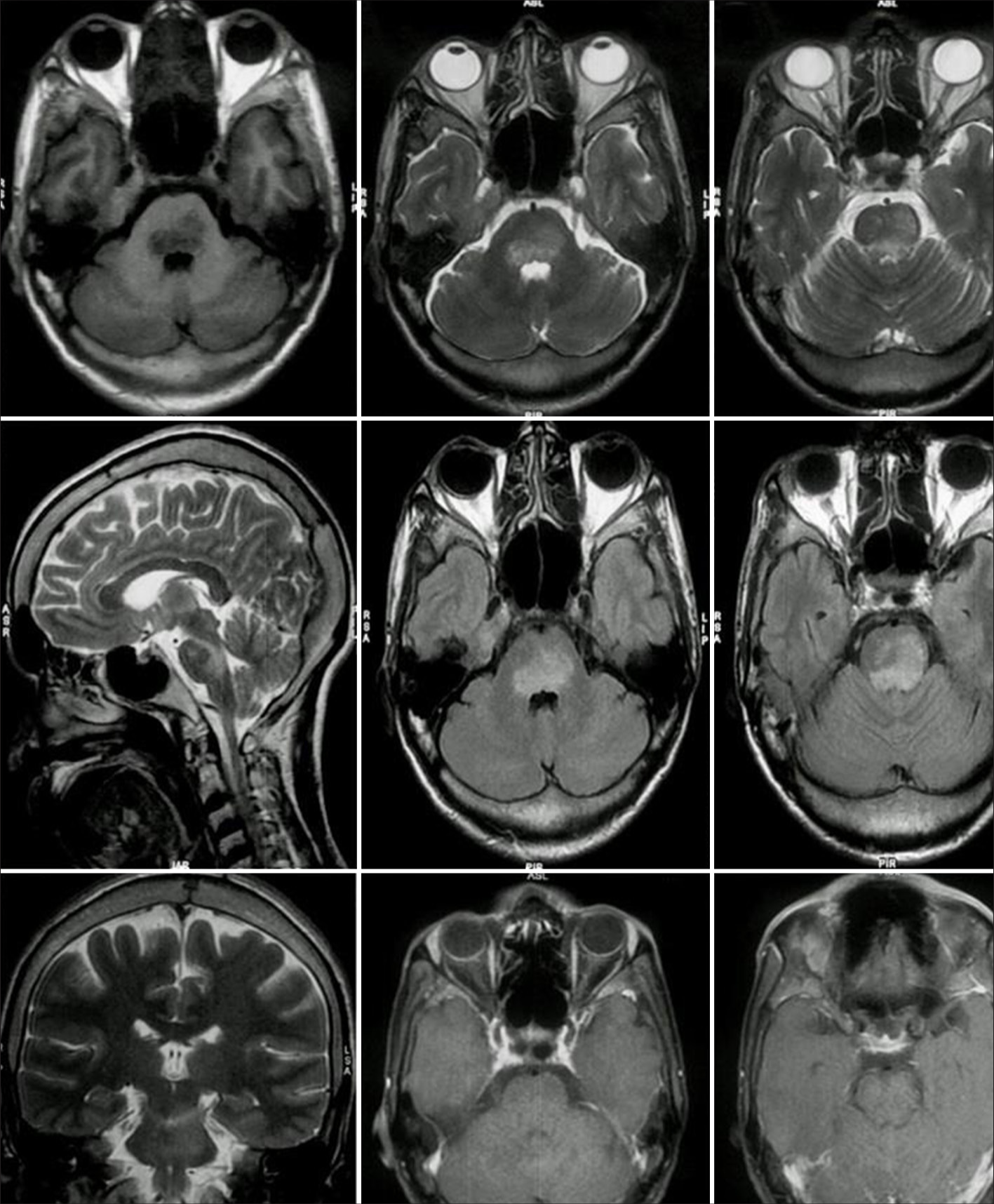
- Magnetic resonance imaging of the brain showing fluffy non-enhancing hyperintensities involving the pontine tegmentum.
A diagnosis of MOGAD presenting as rhombencephalitis with subclinical optic neuritis was determined based on clinical, electrophysiological, and radiological findings, and the patient was managed with intravenous pulse steroid therapy (1 g/day for 5 days). By day 4 of treatment initiation, the patient started showing improvement in horizontal ocular movements and was discharged within a week. The patient recovered completely at two months of follow-up clinically and radiologically without any adverse events [Supplementary video 2].
DISCUSSION
This case covers two important areas in MOGAD, namely presentation as rhombencephalitis, and anatomical localization of horizontal gaze palsy.
The above-mentioned presentation as a core clinical event (brainstem deficit-horizontal gaze palsy) with a positive antibody test points toward the diagnosis of MOGAD as per the 2023 International MOGAD Panel criteria.[3] Horizontal gaze palsy associated with lower motor neuron facial with spared vertical gaze, pupillary reflexes favors localization to abducens nucleus of the pons near the floor of the fourth ventricle which correlates with neuroimaging.[4] The presentation following fever is suggestive of an infective trigger that causes immune response through mechanisms of molecular mimicry, bystander, and polyclonal activation with associated production of MOG reactive B-cells. Brainstem involvement is usually associated with other common MOGAD-related syndromes like optic neuritis which was subclinical with a prolonged visual evoked potential in this case.[5,6] This case shows the importance of suspecting and working up for MOGAD in atypical presentations, prompt identification of which can lead to favorable outcomes in such patients. The strength of this case report lies in the diagnostic work-up and treatment outcome of the rare presentation in MOGAD. The limitation was that a repeat MOGAD antibody was not done at follow-up.
CONCLUSION
MOGAD has a wide spectrum of manifestations spanning from optic neuritis to ADEM and ADEM-like presentations. This case demonstrates such a presentation as rhombencephalitis. Early diagnosis and prompt intervention are crucial to avoid long-term disability in MOGAD.
Ethical approval
Institutional Review Board approval is not required.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship: Nil.
References
- The spectrum of MOG autoantibody-associated demyelinating diseases. Nat Rev Neurol. 2013;9:455-61.
- [CrossRef] [PubMed] [Google Scholar]
- Myelin oligodendrocyte glycoprotein-antibody associated disease: An updated review of the clinical spectrum, pathogenetic mechanisms and therapeutic management. Antibodies (Basel). 2024;13:43.
- [CrossRef] [PubMed] [Google Scholar]
- Diagnosis of myelin oligodendrocyte glycoprotein antibody-associated disease: International MOGAD Panel proposed criteria. Lancet Neurol. 2023;22:268-82.
- [CrossRef] [PubMed] [Google Scholar]
- Neuro-ophthalmology: Ocular motor system In: Bradley and Daroff 's neurology in clinical practice (8th ed). Netherlands: Elsevier; 2021. p. :219.
- [Google Scholar]
- MOG-IgG in NMO and related disorders: A multicenter study of 50 patients. Part 3: Brainstem involvement-frequency, presentation and outcome. J Neuroinflamm. 2016;13:281.
- [CrossRef] [PubMed] [Google Scholar]
- MOG antibody-associated optic neuritis. Eye (Lond). 2024;38:2289-301.
- [CrossRef] [PubMed] [Google Scholar]




