
Translate this page into:
Mononeuritis multiplex in acquired amegakaryocytic thrombocytopenia
Address for correspondence: Dr. Firosh Khan, Department of Neurology, Mother Hospital, Thrissur - 680 012, Kerala, India. E-mail: firoshkhans@gmail.com
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Mononeuritis multiplex involves inflammation of two or more nerves, typically in unrelated parts of the body. It has been well described in bleeding disorders like idiopathic thrombocytopenic purpura (ITP) and Hemophilia. Acquired amegakaryocytic thrombocytopenia (AAT) is a bleeding diathesis characterized by thrombocytopenia but with reduced number of megakaryocytes in the bone marrow, as against ITP. Though AAT is a well described entity, peripheral nervous system manifestations have not been described so far. We report a young man who has presented with bleeding diathesis and mononeuritis multiplex due to AAT. The mechanism of development of mononeuritis multiplex and treatment options are discussed.
Keywords
Amegakaryocytic thrombocytopenia
mononeuritis multiplex
purpura

Introduction
Acquired amegakaryocytic thrombocytopenia (AAT) is a rare disease characterized by immunologically mediated peripheral thrombocytopenia similar to idiopathic thrombocytopenic purpura (ITP). However, in contrast to ITP, there is selective reduction/absence of bone marrow megakaryocytes.[12] Though multiple simultaneous/sequential peripheral nerve involvement (mononeuritis multiplex) is rarely described in ITP,[3] it is hardly ever described in AAT. We are reporting such a case of mononeuritis multiplex due to a cyclical type of AAT in a young man.
Case Report
A 34-year-old man presented in neurology service with pain in the legs, bilateral foot drop and numbness in the medial aspect of the left forearm of 2 weeks duration. Examination showed pupura and on positive questioning he admitted bleeding from gums starting 2 days prior to neurological symptoms. At the age of 12 years, he had multiple blood transfusions and was told to have aplastic anemia, but a detailed medical report was not traceable. At the age of 22 years, he had an acute hepatitis B infection when the blood counts and hematocrit were documented to be normal.
At the time of index admission, he had anemia and multiple purpuric skin lesions [Figure 1]. Systemic examination otherwise was normal. Clinical neurological examination revealed sensory motor involvement of the left radial, left ulnar, right common peroneal and left sciatic nerves suggesting a mononeuritis multiplex. No significant hematomas were seen on the overlying skin of these nerves. An electrophysiology evaluation showed the sensory motor involvement of the left ulnar, left median, left radial, right median, right common peroneal and left sciatic nerves confirming mononeuritis multiplex. There were conduction blocks demonstrable in right peroneal and left ulnar nerves [Figure 2]. After the neurological investigations, he was referred to a Government teaching hospital where his subsequent hematological workup was done.
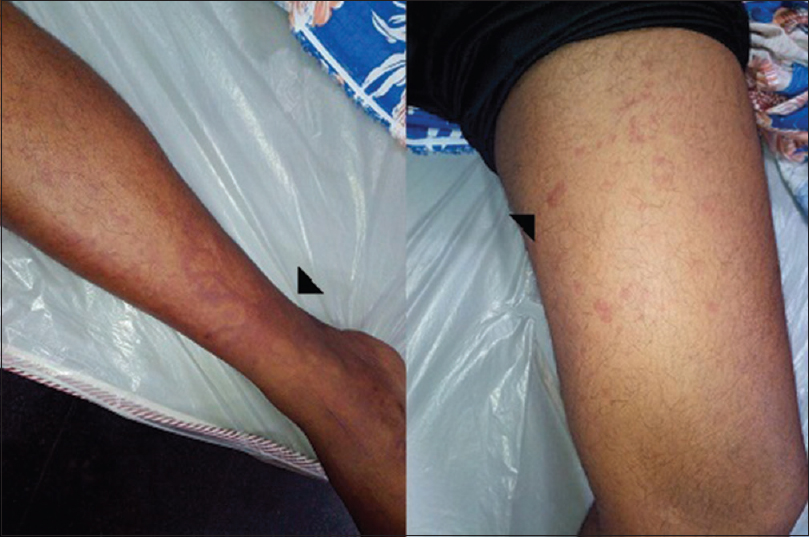
- Echymotic and purpuric skin lesions in legs (arrows)
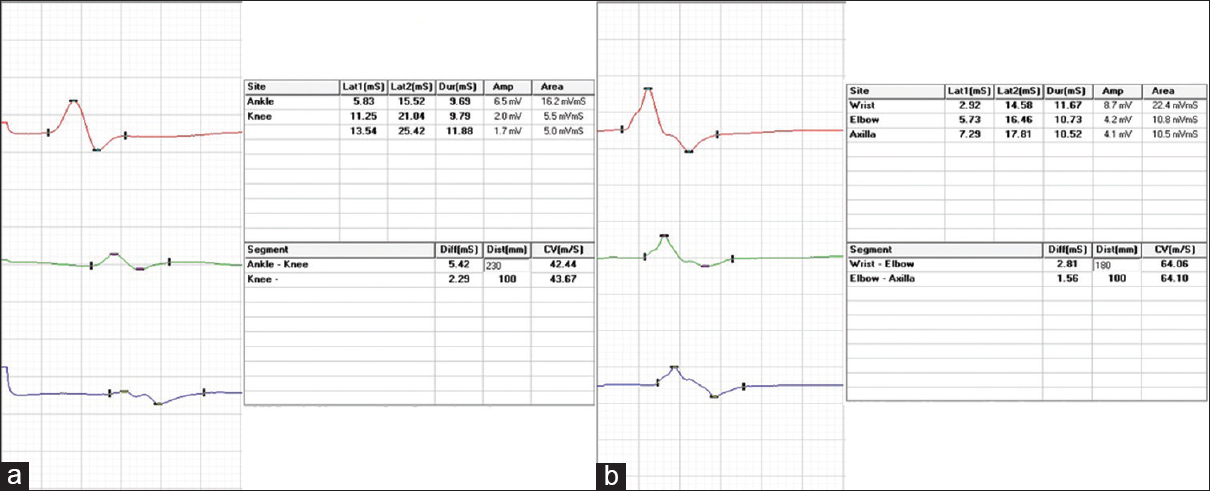
- Motor conduction blocks (more than 50% reduction in compound muscle action potential amplitude with proximal stimulation compared to distal stimulation) in (a) right peroneal nerve and (b) left ulnar nerves
His hematocrit was 33%; total leukocyte count, differential count, and erythrocyte sedimentation rates were normal. The platelet count was grossly reduced to just 5000 cells/cu mm and the bleeding time was prolonged to 9 min. Clotting time, prothrombin time and activated partial thromboplastin time were normal. Corrected reticulocyte count was 2.9% and peripheral blood smear showed marked thrombocytopenia [Figure 3]. The serum bilirubin was marginally elevated to 1.86 mg/dl with the predominant indirect component. Both direct and indirect coomb's tests were negative. Measurements of serum sodium, potassium, calcium, phosphorus, glucose, urea, creatinine, proteins, aspartate aminotransferase and alkaline phosphatase levels were normal. Serum antinuclear antibody, cANCA, pANCA and cryoglobulins were reported negative. Serological tests for HIV, hepatitis B, hepatitis C and syphilis were negative.
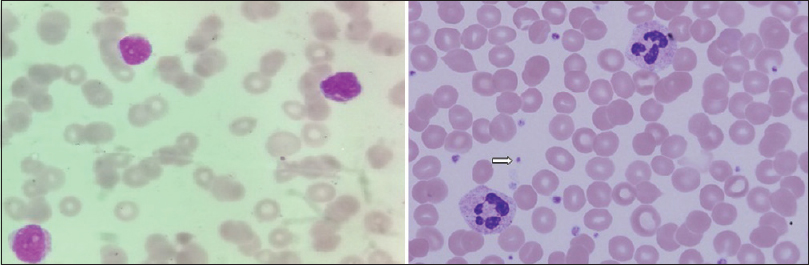
- Peripheral blood smear (Leishmans stain, ×100) showing markedly reduced platelets; compare with normal smear in right panel showing platelets (arrow)
A bone marrow aspiration study, after relative stabilization, showed normoblastic erythroid/myeloid series, normal lymphoid cells and marked reduction of megakaryocytes [Figure 4]. He had profuse bleeding from the site of aspiration with a fall in hematocrit up to 17% and hence a bone marrow trephine biopsy and a nerve biopsy were not attempted. The life-threatening hemorrhage was treated with blood transfusions. He was initiated on oral prednisolone, and since the condition was not improving, later intravenous immunoglobulin was given for 5 days, and cyclosporine A was added. Gradually he stabilized, bleeding stopped and platelet count improved to 60,000 cells/cumm. At the time of hospital discharge at 6 weeks, he had only minimal bilateral foot drop and mild left grip weakness. After 1 week of discharge, he was readmitted with acute onset massive intracerebral hemorrhage, to which he succumbed within few hours.
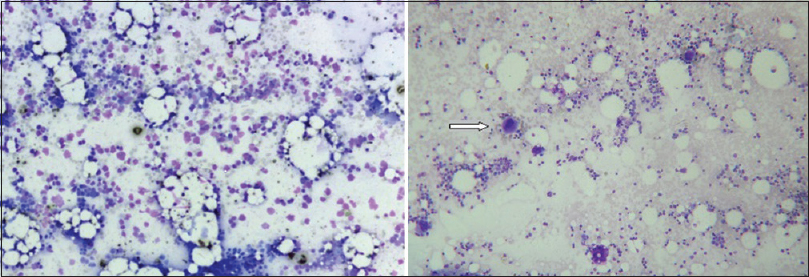
- Absent megakaryocytes in bone-marrow (Leishmans stain, ×10); compare with megakaryocytes of normal bone-marrow in right panel (arrow)
Discussion
We report a case of mononeuritis multiplex due to AAT. Our patient presented with painful bilateral foot drop along with skin lesions. The clinical and electrophysiological features were suggestive of a mononeuritis multiplex since there was asymmetric involvement of multiple peripheral nerves. Skin rashes in mononeuritis multiplex are not rare, and the major differentials include collagen vascular diseases, systemic vasculitides, secondary antiphospholipid antibody syndrome, amyloidosis, sarcoidosis, lyme disease, leprosy and postviral syndromes.[4] Hepatitis B virus infection needs special mention since its long-term autoimmune manifestations like systemic lupus erythematosus, antiphospholipid syndrome, polyarteritis nodosa and rheumatoid arthritis can cause mononeutis multiplex with a skin rash.[5] However, hepatitis B serology was negative in our patient though he had an acute infection previously.
The rashes in our patient were purpuric, and he had severe thrombocytopenia. Detailed evaluation revealed markedly reduced megakaryocytes in the bone marrow with relatively normal erythroid and myeloid series. This hematological picture was consistent with what is described as AAT, as against ITP with similar peripheral blood picture, but normal or increased megakaryocytes in bone marrow.[12] The neuropathy in our patient was probably due to bleeding into the nerves as a result of thrombocytopenia. Similar neuropathy is well described in ITP, where the cause was histologically proven to be intraneural hematoma beneath the epineurium, in between the fascicles.[3] But there is hardly any report on neuropathy in amegakaryocytic thrombocytopenia till date.
Acquired amegakaryocytic thrombocytopenia is a rare disease characterized by severe thrombocytopenia with a total absence or a selective decrease in bone marrow megakaryocytes.[12] It may be a primary disorder or may be seen in aplastic anemia, preleukemia, systemic lupus erythematosus, congenital rubella, dengue fever, ethanol abuse, nutritional B-12 deficiency, radioiodine therapy and thrombocytopenia-absent radius syndrome.[6] A variety of pathogenetic mechanisms were suggested, but an immune-mediated mechanism, due to an autoantibody that blocks the action of endogenous thrombopoietin on megakaryocytopoiesis, appears sensible, at least in a subset of patients.[2] The effectiveness of cyclosporine A in improving thrombocytopenia strongly suggests this mechanism.[7] Our patient also had at least a partial response to immunotherapy initially.
The clinical course of AAT is unpredictable. While some patients remain clinically stable, others may progress to aplastic anemia or myelodysplastic syndrome.[126] A cyclical course has also been described,[8] and our patient appears to be in this category though the hematological picture during the initial episode is not clear. Documented normal hematological picture between the two episodes underscores this.
Congenital amegakaryocytic thrombocytopenia (CAT) is another related rare disease presenting with isolated thrombocytopenia in infancy, developing into pancytopenia in later childhood. As against the immune-mediated mechanism in AAT, defective surface expression of the thrombopoetin receptor c-mpl over hematopoietic progenitor cells due to c-mpl mutations could explain deficient amegakaryocytes in CAT.[9]
Various treatments have been tried in AAT, including corticosteroids, lithium carbonate, androgens, vincristine, folic acid, platelet and erythrocyte transfusions and plasma substitution. An occasional patient may respond well to an intravenous immunoglobulin or cyclosporin A.[7] But, the only real hope is bone-marrow transplantation using a human leukocyte antigen-matched donor.[12]
Acknowledgment
We would like to acknowledge the help of Dr. Sowmini Unnikrishnan MD, Department of Pathology, Lisie Hospital, Cochin, Kerala, India.
Source of Support: Nil.
Conflict of Interest: None declared.
References
- Painful mononeuritis multiplex in idiopathic thrombocytopenic purpura. Indian Pediatr. 2005;42:621-2.
- [Google Scholar]
- Hepatitis B virus (HBV) and autoimmune disease. Clin Rev Allergy Immunol. 2008;34:85-102.
- [Google Scholar]
- Acquired amegakaryocytic thrombocytopenic purpura: A syndrome of diverse etiologies. Blood. 1982;60:1173-8.
- [Google Scholar]
- Successful treatment of amegakaryocytic thrombocytopenic purpura with cyclosporine. N Engl J Med. 1985;312:1060-1.
- [Google Scholar]
- Juvenile cyclic amegakaryocytic thrombocytopenia: A novel entity. J Pediatr Hematol Oncol. 2005;27:148-52.
- [Google Scholar]
- c-mpl mutations are the cause of congenital amegakaryocytic thrombocytopenia. Blood. 2001;97:139-46.
- [Google Scholar]




