
Translate this page into:
Longitudinally Extensive Transverse Myelitis: One Disease, Variable Outcomes—A Case Series

Amit Kumar Satapathy, MD Department of Pediatrics, All India Institute of Medical Sciences Bhubaneswar 751019, Odisha India ped_amit@aiimsbhubaneswar.edu.in
This article was originally published by Thieme Medical and Scientific Publishers Pvt. Ltd. and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Abstract
Longitudinal extensive transverse myelitis (LETM) is a rare form of widespread inflammation of the spinal cord causing T2 hyperintensity in spinal magnetic resonance imaging (MRI) extending across three or more vertebral segments. It is an acute onset of sensory, motor, and autonomic dysfunction of variable etiology with a likely poor outcome. We present a case series of three cases wherein children between the ages of 4 and 13 years had diverse symptoms from gradual painless loss of vision in both eyes with headache, vomiting and seizure, and a normal central nervous system examination except involvement of the optic nerve to another child with abdominal pain, urinary retention and constipation for 3 days with exaggerated DTR, and patchy sensory loss without any definite sensory level, and to the third child with fever and weakness of lower limbs, hypotonia and grade 1–2/5 power in lower limbs and normal upper limb power. Contrast-enhanced MRI spine of all children showed long segment T2 hyperintensity with variable involvement of the brain. The first two children were treated with pulsed dose methylprednisolone, and the last child received intravenous immunoglobulin followed by methylprednisolone. All were followed with oral prednisolone. LETM has a varied presentation with different etiologies. Antineuromyelitis optica immunoglobulin G (IgG) antibody (Aquaporin-4 IgG) and antimyelin oligodendrocyte glycoprotein antibody are strongly recommended though they may not be locally available or not affordable. Early and aggressive immunomodulatory therapy may help faster recovery, as did with two of our three children.
Keywords
paraparesis
Aquaporin-4
myelin oligodendrocyte glycoprotein

Introduction
Childhood acute transverse myelitis (ATM) has an incidence of 1.7 to 2 per million per year and contributes to almost 20% of the demyelinating neurological illnesses in children less than 18 years of age.1 ATM could be either short segment or longitudinally extensive. Longitudinal extensive transverse myelitis (LETM) requires demyelination to involve three or more contiguous vertebral segments.1 2 LETM is often associated with neuromyelitis optica (NMO), but postinfectious and spinal cord infarction are other possibilities. NMO and its spectrum involve the coexistence of inflammatory myelitis and optic neuritis without any symptomatic disease outside these regions and was initially reported in 1870.1 LETM can also be the presenting feature of various autoimmune conditions, such as systemic lupus erythematosus, acute disseminated encephalomyelitis (ADEM), Sjogren's syndrome, may be part of a viral infection, and occasionally even after detailed investigations, no cause may be found.1 The prevalence of NMO spectrum disorders (NMO-SDs) in various studies from the western world ranges from as low as 0.022 in the Caucasian population to as high as 10 per 1,00,000 in the East Asian or black populations.3 4 Early identification of anti-NMO immunoglobulin G (IgG) antibody (Aquaporin-4 [AQP4] IgG) and anti-myelin oligodendrocyte glycoprotein (anti-MOG) antibody helps differentiate this from other autoimmune disorders.
Case 1
A 4-year-old boy presented with gradual painless loss of vision in both eyes with headache, vomiting, and one episode of a generalized tonic-clonic seizure. He had absent light perception in both eyes, with the rest of the systemic examination being normal. There were no other features suggestive of inflammatory connective tissue disease. Laboratory investigation revealed a normal hemogram, erythrocyte sedimentation rate of 15 mm/h, negative C-reactive protein, and negative antinuclear antibodies (ANAs). Ophthalmology examination showed disc pallor in both eyes. Cerebrospinal fluid (CSF) analysis revealed 10 cells, predominantly mononuclear with normal glucose and protein and a negative culture. Contrast-enhanced magnetic resonance imaging (CEMRI) of the brain with spine showed T2 hyperintense signal with minimal enhancement in the cord involving both gray and white matter with mild cord expansion extending from D6–D7 to D10–D11 suggestive of a LETM with the brain being normal (Fig. 1A). Visually evoked potential (VEP) revealed prolonged P100 latency indicative of demyelination. EEG was normal. The child was managed with pulsed injection methylprednisolone for 3 days followed by oral prednisolone for 1 month. The child recovered clinically with 6/6 vision after 7 days of starting treatment with no further seizures. He is on regular follow-up and is doing well.
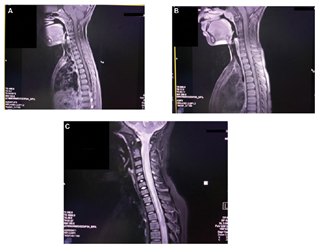
-
Fig. 1 (A) T2 hyperintense signal with minimal enhancement in the cord involving both gray and white matter with mild cord expansion extending from D6–D7 to D10–D11. (B) T2 hyperintensity in the cord opposite D4–D5. (C) T2-hyperintense signals seen in the central aspect of the cord occupying more than two-thirds of the cross-sectional area in cervical and dorsal (cervical > dorsal) regions extending cranially till anterior medulla and caudally till conus medullaris.
Fig. 1 (A) T2 hyperintense signal with minimal enhancement in the cord involving both gray and white matter with mild cord expansion extending from D6–D7 to D10–D11. (B) T2 hyperintensity in the cord opposite D4–D5. (C) T2-hyperintense signals seen in the central aspect of the cord occupying more than two-thirds of the cross-sectional area in cervical and dorsal (cervical > dorsal) regions extending cranially till anterior medulla and caudally till conus medullaris.
Case 2
A 12-year-old boy was admitted with complaints of abdominal pain and distention and loss of bladder and bowel sensations for 3 days. On examination, the child had a distended abdomen with bilateral pitting pedal edema. His higher mental functions, cranial nerve, and fundus examinations were normal. Motor examination showed flaccid lower limbs with a power of 3/5 on all muscle groups. Deep tendon reflexes (DTRs) were sluggish with bilateral upgoing plantar reflexes. A sensory level was identified just below the umbilicus. Bladder and bowel sensations were absent. The VEP was normal. CEMRI of the brain with the spine showed hyperintense signal with patchy enhancement seen in the cervical cord involving both gray and white matter with mild cord expansion extending from C2–C3 to C6–C7. T2 hyperintensity was also noted in the cord opposite D4–D5, suggestive of LETM (Fig. 1B). CSF examination was normal, and ANA, gastric aspirate for CBNAAT (cartridge-based nucleic acid amplification test), and chest radiography were also normal. Anti-NMO IgG antibody (AQP4 IgG) testing could not be done due to financial reasons. The child received pulsed injection methylprednisolone for 3 days followed by oral prednisolone for 1 month. The child improved dramatically and is doing well on follow-up at 1 year after discharge.
Case 3
A 13-year-old boy was brought to us after being treated elsewhere for 20 days for meningoencephalitis. After 4 days of discharge from the hospital, he again developed fever, inability to walk, and loss of bladder and bowel sensation; hence he was brought to us. He had stable vital signs. Central nervous system (CNS) examination revealed higher mental functions and cranial nerve examinations were normal. Motor examination showed him to have grade 1–2 power in all groups of muscles in both lower limbs, brisk DTRs in the lower limbs, and extensor plantar reflexes bilaterally. A sensory level was assessed at T6, and he had no bladder and bowel sensation. Fundus examination showed bilateral papilledema. Other systemic examination was normal. Mannitol was started in view of papilledema, and a CEMRI brain with the spine was done, which showed cervical and thoracic T2-hyperintense signals seen in the central aspect of the cord occupying more than two-thirds of the cross-sectional area in cervical and dorsal (cervical > dorsal) regions extending cranially till anterior medulla and caudally till conus medullaris suggestive of LETM (Fig. 1C). ANA, chest radiography, and anti-NMO IgG antibody (AQP4 IgG) were negative. The child was initially given pulsed injection methylprednisolone for 5 days followed by oral prednisolone, and intravenous immunoglobulin was given at 2 gm/kg on the 2nd and 3rd days of admission. The child had autonomic dysfunction with fluctuating blood pressures and intermittent bradycardia. Despite the above measures initiated, his weakness persisted. He was discharged after 1 month of hospital stay without significant clinical improvement after teaching them home care and physical therapy. He is being regularly followed up and has persisting paraparesis with no bladder or bowel sensations even after over 1 year of discharge from the hospital.
Discussion
LETM is a rare, though clinically important, entity in children. It is important to recognize the condition early because of a high recurrence despite appropriate treatment. Now, LETM is incorporated in the revised diagnostic criteria of NMO. NMO-SDs are demyelinating diseases of the CNS which closely mimic multiple sclerosis (MS). In fact, it was previously thought to be a severe form of it. AQP4 antibody disease has a mean age at onset of approximately 40 years with a high female-to-male ratio as high as 9:1, but MOG antibody disease is common in children with a gender ratio closer to 1:1.4 As this disorder has been studied in adults more than in children, the diagnostic criteria are extrapolated from adults, and there is a lack of uniform guidelines for the management of affected children. The 2015 International Consensus Guidelines for NMO5 for diagnosis of patients with positive AQP4-antibody requires at least one core clinical characteristic and exclusion of alternative diagnosis (e.g., sarcoidosis, neoplastic/paraneoplastic, vascular, chronic infection). The same for those whose AQP4-antibody is negative or unavailable needs a minimum of two core clinical characteristics resulting from one or more clinical attacks with
-
At least any one of optic neuritis/LETM/area postrema syndrome,
-
Dissemination in space (two or more different core clinical characteristics),
-
Fulfillment of additional MRI requirements as applicable, and exclusion of alternative diagnoses as discussed above.
Other than the core clinical characteristics mentioned above, acute brainstem syndrome, symptomatic narcolepsy or acute diencephalic syndrome with NMO-SD, typical diencephalic MRI lesions, or symptomatic cerebral syndrome with NMO-SD-typical brain lesions is considered.6 LETM is radiologically defined as a hyperintense intramedullary signal expanding from the cranial to the caudal direction with the involvement of three or more vertebral body heights.7 The differentiating feature of NMO from other disorders that can present with LETM includes the central cord involvement with sparing of the peripheral part of the cord.7 The antibody screen (IgG against AQP4) has a very high specificity (> 99%) with a sensitivity of around 70 to 80%8 but may not necessarily be positive in the early phase of the disease. The mortality rates are high when the cervical lesions extend into the brainstem. Treatment involves the early institution of steroid therapy. All our children satisfied the clinical criteria for LETM though the AQP4 IgG antibody was negative in the one tested. ADEM and MS were ruled out with the absence of brain lesions. There was no vaccination in the period before the onset of the disease in any of our children, ruling out vaccination-induced transverse myelitis. A negative ANA in all three patients made autoimmune causes highly unlikely.
NMO has a variable course and usually presents as a monophasic illness in children. There was a good response to steroids in two of our three children, with one child not recovering after treatment, indicating a variable degree of recovery seen with this illness.9 Therapeutic plasma exchange is an option in children, refractory to the first line of treatment.10 There is no specific defined therapeutic window that has been ascertained, and the adverse events following plasma exchange are minimal, with improvement in functional outcomes.10 This case series is done to improve the awareness of this disorder among pediatricians who are the first-contact physicians. Although there are standard diagnostic criteria available for NMO-SD, the absence of testing facilities poses a diagnostic challenge. As MRI features are very classical and readily available, children with typical imaging findings should be promptly treated without delay. With an increased number of cases being reported from different parts of the country, it calls for appropriate diagnostic criteria and treatment guidelines specifically directed to children.
Conflict of Interest
None declared.
Funding None.
References
- Acute transverse myelitis in children, literature review. Iran J Child Neurol. 2018;12(2):7-16.
- [Google Scholar]
- The demographic, clinical, and magnetic resonance imaging (MRI) features of transverse myelitis in children. J Child Neurol. 2012;27(1):11-21.
- [Google Scholar]
- Epidemiology of neuromyelitis optica spectrum disorder and its prevalence and incidence worldwide. Front Neurol. 2020;11:501.
- [Google Scholar]
- International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. 2015;85(2):177-189.
- [Google Scholar]
- MRI characteristics of neuromyelitis optica spectrum disorder: an international update. Neurology. 2015;84(11):1165-1173.
- [Google Scholar]
- AQP4 antibody-positive Thai cases: clinical features and diagnostic problems. Neurology. 2011;77(9):827-834.
- [Google Scholar]
- Clinical characteristics, course and prognosis of relapsing Devic's neuromyelitis optica. J Neurol. 2004;251(1):47-52.
- [Google Scholar]
- Prognostic indicators of improvement with therapeutic plasma exchange in pediatric demyelination. Neurology. 2019;93(22):e2065-e2073.
- [Google Scholar]




