
Translate this page into:
Late-Onset Lennox-Gastaut Syndrome with Chromosome 15q Duplication in Sisters
 , Adriane Cristina Vieira dos Santos2, Felipe Luan Lima da Silva2, Renata Carvalho Cremaschi1, Fernando Morgadinho Coelho1,3
, Adriane Cristina Vieira dos Santos2, Felipe Luan Lima da Silva2, Renata Carvalho Cremaschi1, Fernando Morgadinho Coelho1,3
Marcos Manoel Honorato, PhD Rosa Vermelha, 2272, Santarém-PA 68010-200 Brazil marcosmhonorato@hotmail.com
This article was originally published by Thieme Medical and Scientific Publishers Pvt. Ltd. and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Abstract
Our study reports two nontwin sisters with late-onset Lennox-Gastaut syndrome and chromosome 15q duplication, showing the evolution, symptoms, diagnosis, and treatment of these patients, with the aim of increasing knowledge about this extremely rare association. They had a variety of generalized seizures types, intellectual disability, electroencephalogram with generalized epileptiform discharges less than 3 Hz, dysmorphisms, and genetic studies with the presence of duplicated chromosome 15. Cases reported here may be related to chromosomal changes inherited from their asymptomatic mother.
Keywords
15q duplication syndrome
epilepsy
genetic
Lennox-Gastaut syndrome

Introduction
Lennox-Gastaut syndrome (LGS) is an epileptic encephalopathy that usually starts in childhood, with cases after 8 years of age being uncommon.1 2 Chromosomopathies are rarely associated with LGS.3 However, 15q duplication syndrome is caused by the presence of at least one extra maternal copy of the Prader–Willi/Angelman syndrome critical region on chromosome 15q11.2-q13.1.4 Its association with late-onset LGS has been rarely described in the literature and usually occurs due to de novo mutation.5 6
Case Report
Patient 1
A 25-year-old female, uneventful in the prenatal, neonatal, and perinatal periods, presented with delay in psychomotor development, gait and speech at the age of 2 years. She is the daughter of nonconsanguineous parents and a healthy mother.
She started mood changes and insomnia at the age of 18 years. Months later, it evolved with the first tonic–clonic generalized seizures, then frequent prolonged absences and atonic seizures. There were cognitive worsening and periods of unresponsiveness. In adulthood, they remain disoriented, with dysarthria, apraxia, and ataxia. It also presents dysmorphism with small hands, in addition to palmar and adductor pollicis tendon retraction (Fig. 1).

-
Fig. 1 Unprecedented dysmorphism found in both sisters: small hands, in addition to palmar and adductor pollicis tendon retraction.
Fig. 1 Unprecedented dysmorphism found in both sisters: small hands, in addition to palmar and adductor pollicis tendon retraction.
Magnetic resonance image did not detect abnormalities and electroencephalogram (EEG) revealed disorganized baseline brain activity and frequent epileptiform generalized spike-wave discharges. 1.5 to 2 Hz. (Fig. 2).
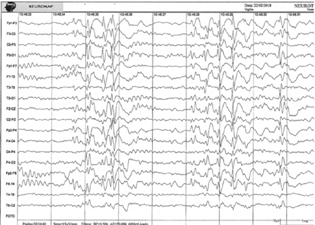
-
Fig. 2 EEG fragment from patient 1 showing diffuse disorganization of the baseline brain electrical activity associated with generalized epileptiform spike-wave discharges at 1-2 Hz.
Fig. 2 EEG fragment from patient 1 showing diffuse disorganization of the baseline brain electrical activity associated with generalized epileptiform spike-wave discharges at 1-2 Hz.
She currently uses divalproex, nitrazepam, levetiracetam, and lamotrigine, with partial control. A genetic test was performed (epilepsy panel) with a diagnosis of 15q duplication that extends at least 1.434 Mb from Chr15:25,584,287 through Chr15:27.018.935, and involves two genes previously associated with epilepsy: UBE3A and GABRB3.
Patient 2
A 27-year-old female, uneventful in the prenatal, perinatal, and neonatal periods, presented with neuropsychomotor development delay, with gait at the age of 2 years and speech at the age of 3 years, had learning difficulties, was dependent for activities of daily living. In childhood, he evolved with aggressive behavior.
Her epileptic seizures started at the age of 2 years, which occurred at intervals of a few months, but after 12 years of age they became more intense, occurring several times a day with atonic, tonic, myoclonic, and absences seizures.
She speaks a few, without answering questions, obeys few commands (apraxia), with dysarthria, occasional echolalia, appendicular, and gait ataxia. In addition, it has dysmorphisms: small hands with bilateral palmar and adductor pollicis tendon retraction.
Currently, she uses combination of divalproex, lamotrigine, levetiracetam, and nitrazepam. She achieved a 70% reduction in seizures, as well as an improvement in behavior and aggressiveness.
Skull computed tomography did not show abnormalities and EEG detected diffusely disorganized baseline activity, with predominance of theta waves and presence of frequent generalized epileptiform activity with a frequency around 1.5 Hz and high voltage. Genetic study detected the same change as her sister, which consists of a chromosome 15 duplication.
Discussion
LGS is an epileptic encephalopathy described in childhood usually between 1 and 7 years.7 It is characterized by several types of generalized epileptic seizures, with poor response to antiseizure medication associated with intellectual delay and behavior disorders, and it presents a specific EEG pattern with epileptiform discharges <3.0 Hz.8 There are a variety of etiologies and the chromosome 15q duplication has been linked among them.5 The case series demonstrates a mutation in the same offspring with different gene expression; however, culminating in late-onset LGS, which is uncommon both due to the age of onset and the chromosomal etiology, is rare. On 15q duplication, individuals with idic (15) are typically more severely affected than those with interstitial duplication. In idic (15), the penetrance is 100% and the expressiveness is variable. De novo mutations were reported and the risk for siblings is low, but it is assumed to be marginally higher than in the general population, due to the possibility of germline mosaicism.4
However, the maternal interstitial duplication of 15q11.2-q13.1 was de novo in 85% of the probands and inherited from the mother in 15%. Thus, if the mother has the 15q interstitial duplication, the risk of each child inheriting the duplication is 50%. In the maternal interstitial duplication of 15q11.2-q13.1, although penetration appears to be complete, some individuals may have mild features that appear to be unaffected, reflecting variable expressiveness rather than true penetration.4
The reported case series possibly demonstrates an interstitial duplication when considering the genetic probability of affecting two individuals of the same offspring, which could represent the first case of LGS associated with interstitial duplication with chromosomal alterations inherited from an asymptomatic mother. Unfortunately, it was not possible to carry out genetic research on the mother. Unfortunately, it was not possible to submit her to genetic research.
The description of several nonspecific dysmorphic characteristics associated with the mutation, such as slanted palpebral fissures, epicanthal folds, microcephaly and extremity anomalies such as clinodactyly and syndactyly of the second and third fingers, among others, has already been described by other authors.9 However, such dysmorphic features were not found in our cases, and the hand dysmorphism of the two sisters was not described in previous studies, suggesting a previously unknown variant.
Conclusions
The reported cases open the possibility of a new variant of the chromosome 15q duplication syndrome, possibly of asymptomatic maternal origin. Establishing the diagnosis, despite not modifying the evolution of the disease so far, allows to guide the forms of future treatment by safely delimiting the causative mechanisms of the disease. In addition, the diagnosis is of fundamental importance to promote genetic counseling.
Conflict of Interest
None declared.
Funding None.
References
- Definition and natural history of Lennox-Gastaut syndrome. Epilepsia. 2011;52(05):3-9.
- [Google Scholar]
- Maternal 15q Duplication Syndrome. 1993. In GeneReviews®, University of Washington, Seattle, Seattle (WA);
- [Google Scholar]
- Late-onset Lennox-Gastaut syndrome as a phenotype of 15q11.1q13.3 duplication. Epileptic Disord. 2012;14(2):159-162.
- [Google Scholar]
- Late-onset Lennox-Gastaut syndrome in a patient with 15q11.2-q13.1 duplication. Am J Med Genet A. 2009;149A(5):1033-1035.
- [Google Scholar]
- [Development of Lennox-Gastaut syndrome in adulthood] Rev Neurol. 2011;52(5):257-263.
- [Google Scholar]
- Lennox-Gastaut syndrome: a state of the art review. Neuropediatrics. 2017;48(3):143-151.
- [Google Scholar]
- Duplication of the 15q11-q13 region: clinical and genetic study of 30 new cases. Eur J Med Genet. 2014;57(1):5-14.
- [Google Scholar]




