
Translate this page into:
Late-onset Familial Amyloidotic Polyneuropathy with Bence Jones Proteinuria and Cardiomyopathy
Address for correspondence: Dr. Sira Carrasco García de León, Department of Neurology, Hospital General Universitario Ciudad Real, C/O Bispo Rafael Torija, s/n 13005, Ciudad Real, Spain. E-mail: siracarrasco@hotmail.com, siracarrasco79@gmail.com
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Familial amyloidotic polyneuropathy is a genetically determined disease characterized by deposition of an anomalous transthyretin. A high index of suspicion is needed for this multisymptomatic and lethal disease to be diagnosed. The patient was a 70-year-old male examined due to hypesthesia in the hands and feet, plus difficulty walking. A neurophysiological study delivered the diagnosis of axonal sensorimotor polyneuropathy. He later developed cardiac symptoms and diarrhea. Urine laboratory analyses revealed a monoclonal spike of light chains (kappa). Biopsies of abdominal fat and bone marrow yielded normal results. The genetic study was compatible with a heterozygous Val30Met-transthyretin mutation. Very few case studies have described an association between familial amyloidotic polyneuropathy and monoclonal gammopathy. We stress that genetic confirmation is important regardless of the type of amyloid deposition revealed by the biopsy.
Keywords
Bence Jones proteinuria
cardiac amyloidosis
transthyretin amyloidosis

Amyloidosis refers to a heterogeneous group of illnesses whose pathogenic mechanism is characterized by extracellular deposition of insoluble protein fibers that will affect the normal architecture and function of organs and tissues.[1] The systemic variants include primary amyloidosis and familial amyloidosis. The former has to do with the presence of lymphoproliferative neoplasm that produces monoclonal light chains; the latter arises from mutations affecting TTR.[2] It is even regarded as its own nosological entity: Familial amyloidotic polyneuropathy or Corino Andrade disease.[2] We present the case of a patient who experienced late-onset sensorimotor polyneuropathy that was predominantly axonal and progressed quickly. This case of polyneuropathy is peculiar because it was associated with two potential causes of amyloidosis: Hypogammaglobulinemia with Bence Jones (kappa) proteinuria and a TTR gene mutation. We stress that genetic testing is important regardless of the type of amyloid deposition revealed by the biopsy.
A 70-year-old man presented with a personal history of type 2 diabetes and former alcohol dependency (wine, moderate). He had been diagnosed with erectile dysfunction in 2009 and was in follow-up by the Internal Medicine Department due to constitutional symptoms. His mother had died suddenly at the age of 68, and two of his sisters had heart failure. He made an appointment in 2013 because of a 1-year history of tingling and hypesthesia in his hands and feet. In the preceding 6 months, he had experienced gradual loss of strength in all four limbs, as well as difficulty walking. The physical examination showed cutaneous ulcers on both feet [Figure 1]. The neurological examination revealed cachexia with marked atrophy of the quadriceps, calf, and forearm muscles and of the interossei and thenar eminences in both hands [Figure 2]. Muscle strength in the upper limbs was rated as follows: Deltoids 4/5, elbow flexion-extension (biceps and triceps) 4/5, wrist flexion-extension 3/5, finger flexion-extension 2/5. Muscle strength in the lower limbs was rated as follows: Thigh flexion (iliopsoas) 4/5, knee flexion-extension (quadriceps and ischiotibial muscles, biceps, semitendinosus and semimembranosus muscles) 4/5, and foot dorsiflexion 3/5. Universal areflexia was observed. Other findings included glove-and-stocking hypesthesia, along with severe and bilateral impairment of vibratory sensation and arthrokinetic sensitivity in the upper and lower limbs.
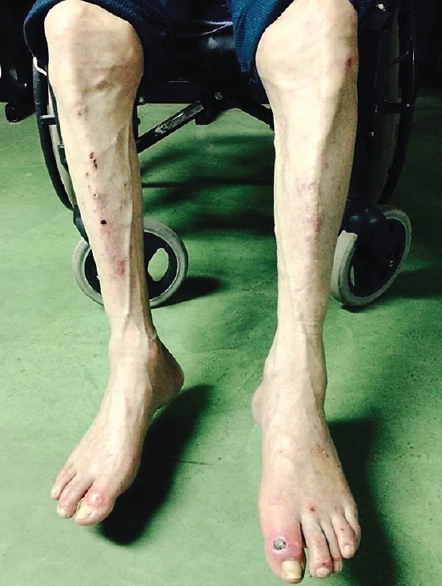
- Diffuse amyotrophy in lower limbs. Neuropathic cutaneous ulcers located on the great toe of each foot
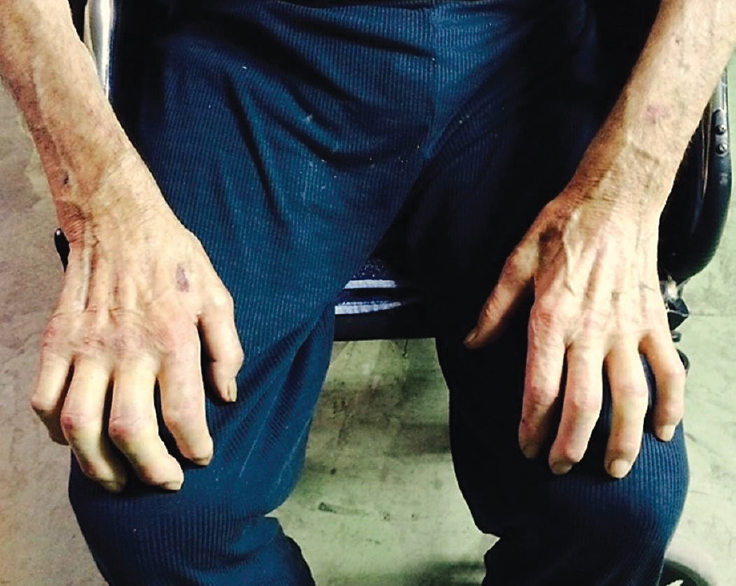
- Amyotrophy of the dorsal interossei of both hands and tendency toward ulnar claw
Laboratory analyses yielded normal erythrocyte sedimentation rate and levels of thyroid hormones and vitamin. Glycosylated hemoglobin (HbA1c) was 6.50% (normal range: <7). The patient tested negative for tumor markers and onconeural antibodies; serology studies were also negative. Immunoelectrophoresis revealed hypogammaglobulinemia with Bence Jones kappa proteinuria of 3.69 (normal range: <1.85). An electromyogram showed a lack of distal evoked responses at both common peroneal nerves, the posterior tibial nerve, and the right median nerve; severely diminished amplitude in the left median and both cubital nerves; and moderately diminished motor conduction speed in all upper limb nerves subjected to the study. The study results were compatible with sensorimotor polyneuropathy that was mixed-type although predominantly axonal.
In light of these general characteristics, we ordered an endoscopy as well as a thoracic, abdominal, and pelvic computed tomography scan; both yielded normal results. Lumbar puncture delivered normal spinal fluid, culture results were normal, and there were no abnormal cytology findings. Three consecutive bone marrow biopsies indicated monoclonal restriction to kappa light chains with no evidence of amyloid deposition. The previous year (January 2014), the patient visited the Emergency Department with symptoms of dyspnea with mild exertion and subacute onset of edema of the lower limbs. A transthoracic echocardiogram [Figure 3] detected thickening of the ventricular walls, atrioventricular and aortic valves, and interatrial septum. There was also an increase in refringence, suggesting the presence of infiltrative heart disease with moderate overall dysfunction. Given these findings, and an initial suspicion of primary amyloidosis, a heart magnetic resonance imaging scan and abdominal fat biopsy were performed; these did not show amyloid deposits.
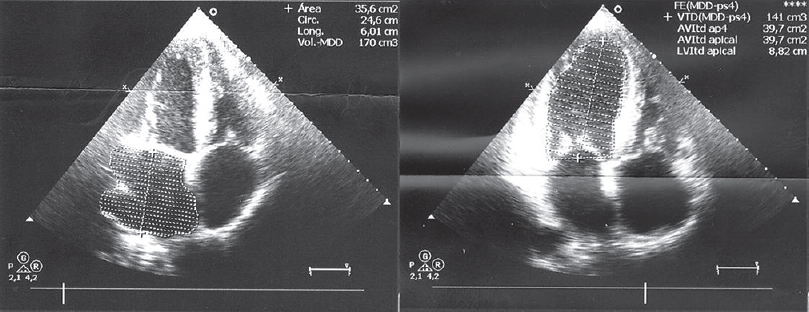
- Transthoracic echocardiogram: Left ventricle with mild thickening of the ventricular wall, except for the basal ventricular septum, which is moderately hypertrophied. Ultrasound findings are compatible with infiltrative cardiomyopathy
These symptoms were accompanied by persistent watery diarrhea. Progressive neurological deterioration resulted in the patient's death in December 2015. A genetic study identified an adenine-guanine substitution at position 30 of exon 2 of the TTR gene; the mutation was heterozygous. This mutation caused valine to be substituted by methionine (Val30Met, c.148G>A, p. Val50Met) as has been described previously.[3] We offered a genetic study to any interested family members aged 18 or older. Nine of the index patient's relatives were studied and three were found to be carriers of the same genetic defect.
Primary amyloidosis is the second most frequent hematological condition associated with a monoclonal protein.[4] Symptoms depend on the organs affected by the deposits and may include myocardiopathy, liver disease, kidney failure, lingual hypertrophy, and polyneuropathy. The most common form of hereditary amyloidosis is the autosomal dominant TTR-associated variant; one of its typical phenotypes is familial amyloid polyneuropathy (FAP).[2] FAP arises due to a single base or point mutation on one allele (heterozygous) or both alleles (homozygous) of the TTR gene located on chromosome 18.[5] The typical presentation includes mixed sensorimotor polyneuropathy that is predominantly axonal with onset in the lower limbs, restrictive cardiomyopathy, dysautonomia, vitreous opacities, diarrhea, impotence, weight loss, and more rarely, leptomeningeal impairment. The classic polyneuropathy syndrome presents with dysautonomia and loss of sensitivity and/or spontaneous pain in the feet due to small fiber impairment.[5]
Diagnosis in the later stages of the disease, as in our case, is more common in patients who have no known family history, mutations outside of the classic nuclear locations (where penetrance is low), and more severe cardiac impairment.[5] We should point out that these late-onset cases are even more difficult to diagnose because a biopsy is less likely to reveal typical amyloid deposits.[6] Our patient also presented other factors adding to the initial diagnostic confusion, including two other potential causes of polyneuropathy: Diabetes mellitus and alcohol dependency. Nevertheless, the disease's inevitable clinical progression (patients are bed- and chair-ridden by 2 years after onset of the polyneuropathic syndrome) did not resemble what would be expected for either of the causes listed above. Lack of response to immunoglobulins, presence of dysautonomias (diarrhea, impotence), and the infiltrative hypertrophic myocardiopathy led us to suspect amyloidosis that initially seemed to be acquired. Confirming hypogammaglobulinemia with kappa light chain proteinuria supported this premise and ruled out the possibility of Bence Jones myeloma; this entity should always be considered given that the literature includes various cases of neuropathy in primary systemic amyloidosis even with no increases in immunoglobulins in plasma and in urine.[7] Nevertheless, the clinical profile was unusual for AL amyloidosis, progression was slow, and the biopsies yielded negative results. In any case, negative biopsies do not rule out a diagnosis of amyloidosis since deposits are frequently patchy.[2] Based on these results, it was suspected an alternative diagnosis and we decided to perform the genetic study that confirmed the presence of a TTR mutation.
This hereditary degenerative disease presents with multiple neurological and nonneurological symptoms that can result in confusion with many other entities. The presence of low-grade monoclonal gammopathy in association with FAP, as in our case, has been described in 24% of all patients.[48910]
The case we describe underscores the importance of suspecting this entity in patients showing axonal polyneuropathy with more marked impairment of thermoalgesic sensitivity and associated dysautonomia and/or infiltrative myocardiopathy, even when there is no relevant family history or when age of onset is advanced. We stress that a study of the TTR gene is necessary regardless of the type of amyloid deposits revealed by a biopsy and even if the patient has comorbidities that might cause polyneuropathy.
Considering that this disease will have a major impact on the lives of the patient and family members, an early diagnosis is crucial for making important treatment decisions. It introduces the possibility of using disease-modifying treatment or integral multidisciplinary treatment, and of avoiding inappropriate and aggressive therapies (such as chemotherapy for AL amyloidosis). It may also enable detection of carriers or cases in early stages.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- Transthyretin (ATTR) amyloidosis: Clinical spectrum, molecular pathogenesis and disease-modifying treatments. J Neurol Neurosurg Psychiatry. 2015;86:1036-43.
- [Google Scholar]
- Amyloid fibril protein in familial amyloidotic polyneuropathy, Portuguese type. Definition of molecular abnormality in transthyretin (prealbumin) J Clin Invest. 1984;74:104-19.
- [Google Scholar]
- Misdiagnosis of hereditary amyloidosis as AL (primary) amyloidosis. N Engl J Med. 2002;346:1786-91.
- [Google Scholar]
- Update in the diagnosis and management of transthyretin familial amyloid polyneuropathy. J Neurol. 2014;261:1227-33.
- [Google Scholar]
- Diagnostic hallmarks and pitfalls in late-onset progressive transthyretin-related amyloid-neuropathy. J Neurol. 2013;260:3093-108.
- [Google Scholar]
- Primary AL (kappa-light chain) amyloidosis manifesting as peripheral neuropathy in young male without increase in serum and urine immunoglobin load: A diagnostic challenge. Clin Neuropathol. 2005;24:118-25.
- [Google Scholar]
- Cardiac amyloidosis, a monoclonal gammopathy and a potentially misleading mutation. Nat Clin Pract Cardiovasc Med. 2009;6:128-33.
- [Google Scholar]
- A sporadic case of late-onset familial amyloid polyneuropathy with Bence-Jones proteinuria. J Neurol. 1999;246:726-7.
- [Google Scholar]
- A sporadic case of late-onset familial amyloid polyneuropathy with a monoclonal gammopathy. Neuromuscul Disord. 2015;25:658-60.
- [Google Scholar]




