
Translate this page into:
Immune-mediated Neuropathies Our Experience over 3 Years
Address for correspondence: Prof. Sadanandavalli Retnaswami Chandra, Faculty Block, Neurocentre, National Institute of Mental Health and Neurosciences, Bengaluru - 560 029, Karnataka, India. E-mail: drchandrasasi@yahoo.com
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Introduction:
Immune-mediated peripheral neuropathy is the term applied to a spectrum of peripheral nerve disorders where immune dysregulation plays a role. Therefore, they are treatable. We analyzed the cases seen in the past 3 years by us and evaluated the clinical, laboratory, and outcome parameters in these patients.
Patients and Methods:
Consecutive patients seen by the authors and diagnosed as immune-mediated neuropathy were analyzed for etiology, pathology, and outcome assessed.
Results:
A total of sixty patients, 31 acute and 29 chronic neuropathies, were identified. Their subtypes treatment and outcome assessed. Males were significantly more in both acute and chronic cases. Miller Fisher 4, AMAN 1, paraplegic type 1, motor dominant type 19, Sensory-motor 1, MADSAM 3, Bifacial 2. Nonsystemic vasculitis was seen in 16 out of 29 chronic neuropathy and HIV, POEMS, and diabetes mellitus one each.
Discussion:
There is a spectrum of immune-mediated neuropathy which varies in clinical course, response to treatment, etc., Small percentage of uncommon cases are seen. In this group, mortality was nil and morbidity was minimal.
Conclusion:
Immune-mediated neuropathies are treatable and hence should be diagnosed early for good quality outcome.
Keywords
Acute inflammatory demyelinating polyneuropathy
chronic inflammatory demyelinating neuropathy
criteria
POEMS syndrome

INTRODUCTION
Immune-mediated disorders of peripheral nervous system form a group of disorders where immune dysregulation plays an important role in disorders affecting peripheral nerves. It is important to recognize them as they are potentially treatable if diagnosed early. Peripheral nervous system is the part of nervous system constituted by cranial nerves 2–12: Roots, plexuses, and nerves. Each nerve contains nerve fascicle, myelin, Schwan cells, fibrocytes collagen, and blood vessels. The disorders that predominantly affect myelin constitute demyelinating disorders, which affect nerve cell body become neuranopathy, which affect axons is axonopathy and vasculitic neuropathy when vasa nervorum are affected and mixed affecting all in various combinations. They can be acute, chronic, relapsing-remitting, etc. The common syndromes included in this group are Guillain–Barre syndrome (GBS) and variants, chronic inflammatory demyelinating neuropathy (CIDP) and variants, motor neuropathy with multifocal conduction block (MMN), vasculitic neuropathy, sarcoid, paraproteinemic neuropathy, and miscellaneous.[12] They are called acute if duration of progression is < 4 weeks, subacute if up to 8 weeks, and chronic if >8 weeks. A 3-6-10 approach is suggested by Sapersten et al., with three goals, six questions, and ten patterns.[3] The goals are to know the cause, site, and plan treatment. Questions are what system in the nerve is affected that is motor, sensory, autonomic or mixed, distribution pattern analysis of nature of sensory involvement, upper motor neuron affected or not, temporal evolution, and heredity. The patterns are proximal and distal symmetrical, distal sensory, asymmetrical distal with sensory, asymmetrical proximal and distal sensory, asymmetrical distal motor, symmetrical distal sensory with upper motor neuron involvement, symmetrical weakness, focal midline proximal, asymmetrical with proprioceptive loss, and those with autonomic nervous system features.
Guillain–Barre syndrome
It is the most common acute type of immune-mediated peripheral neuropathy, generally monophasic and rarely recurrent, with several subtypes. Incidence is 1–2/lakh population. The different types are acute inflammatory demyelinating polyneuropathy (AIDP), acute motor axonal polyneuropathy (AMAN), Acute motor sensory axonal polyneuropathy, Miller Fisher variant, focal variants such as cervico brachio pharyngeal syndromes, acute pandysautonomia, sensory variant, etc., often precipitated by infections, vaccines, sera, surgery secondary to immune-mediated break down of blood-nerve barrier, etc., CD3+ T-cells and macrophages are often seen. Activated complement and membrane attack complexes are also seen. Antibody against major gangliosides, GM1, GD1a, GD1ab, GaLNAc-, are seen in AMAN variant. GQ1Bb in Miller Fisher variant and cause damage probably by molecular mimicry.
Clinical features
Pain, paresthesias, and weakness, autonomic dysfunctions, and cranial nerve palsy especially seventh nerve are common. Mortality is 5%–10% and morbidity is about 20%–30%.[4] The patterns are based on fiber type as motor, sensory, autonomic and mixed, topography as cervicofacial, paraparetic, Miller Fisher and unusual forms, course as monophasic versus relapsing, and based on pathology as axonal versus demyelinating. Treatment is supportive and disease-modifying drugs such as intravenous immunoglobulin 0.4 g/kg/day for 5 days or plasma exchange. Mycophenolate mofetil 2000 g/day and methyl prednisolone for 6 weeks also tried.[5] Eculizumab, a monoclonal antibody, is tried recently in view of its efficacy in preventing complement activation.[6]
Criteria
NINDS critertia: Features required for the diagnosis[7]
Progressive motor weakness of more than one limb, areflexia.
Features strongly supportive of the diagnosis
-
Clinical features
Short progression, relative symmetry, mild sensory symptoms or signs, and cranial nerve involvement. Recovery, autonomic dysfunction, and absence of fever at the onset.
-
Cerebrospinal fluid features
-
Electrodiagnostic features.
Features casting doubt on the diagnosis
Marked persistent asymmetry of weakness, Persistent bladder, or bowel dysfunction, Bladder or bowel dysfunction at onset. More than 50 cells/mm3 in cerebrospinal fluid (CSF), presence of polymorphonuclear cells in CSF, Sharp sensory level.
Features that rule out the diagnosis
A current history of hexacarbon abuse, abnormal porphyrin metabolism, recent diphtheria, and lead neuropathy.
A purely sensory syndrome
Diagnosis of botulism, poliomyelitis, myasthenia gravis, or toxic neuropathy.
Treatment
Plasma exchange, immunoglobulins, and supportive care.[8]
Chronic inflammatory demyelinating neuropathy
A chronic distal and proximal sensory-motor polyneuropathy has a very variable course. Often it presents as fairly symmetrical, sensory motor, distal and proximal with rarely cranial nerve palsy and progresses beyond 8 weeks, unlike GBS. More than fifteen diagnostic criteria are there with less sensitivity but relatively good specificity.[9] Austin in 1958 classified them into motoneuropathy with multifocal conduction block (MMN), multifocal acquired demyelinating sensory and motor neuropathy (MADSAM) (Lewis-Sumner disease), distal neuropathy with IgM, IgA, IgG, anti-MAG paraproteinemia, sensory predominant type, and polyneuropathy, organomegaly, endocrine dysfunction, monoclonal band, skin changes (POEMS) syndrome.[10] CIDP associated with vasculitis purely neurological or with systemic vasculitis, it can be with HIV, hereditary neuropathy, and also diabetes mellitus.
CSF protein is elevated and sural nerve biopsy shows patchy demyelination and mononuclear infiltrates. Steroids, IVIg, and plasma exchange are all used in a case-based way. Azathioprine and cyclophosphamide are used for maintenance therapy.
Criteria
For CIDP, there are more than 15 diagnostic criteria. The parameters included are clinical, electrophysiological, and others such as CSF and biopsy. Commonly American academy of neurology criteria is used.[11]
Classical CIDP is sensory-motor distal and proximal with features of patchy demyelination, axonopathy, and inflammation. CSF shows elevated proteins especially IgG.
Distal-acquired demyelinating symmetric shows symmetrical distal, elevated IgGk, and anti-Gm1 antibody negative.
-
MADSAM, asymmetrical, proximal, multifocal, demyelination common, and CSF often normal.
-
MMN asymmetrical upper limb, distal nerve pattern, anti-GM1 antibody occasionally present.
Multifocal motor neuropathy[12]
Distal muscle weakness, which is purely sensory, is seen. Upper limbs more affected than lower. Males more than females with wasting and fasciculations are seen. GM1, GD1a, and GD1b are commonly associated. As it is purely motor it resembles motor neuron disease. Axonal degeneration with electrophysiological conduction block is seen. Steroids and plasma exchange are not useful. IVIg gives partial response and mostly requires long-term maintenance. Cyclophosphamide, mycophenolate, monoclonal, and anti-CD20 give variable reports.
Polyneuropathy associated with monoclonal gammapathy[13]
About 10% of neuropathy fall in this category. Monoclonal gammopathy of unknown significance, Waldenstrom gammopathy, POEMS, IgM and paraprotein-associated neuropathy. Rarely, IgG- and IgA-associated syndromes are seen.
POEMS syndrome is the term applied to a syndrome of polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes. Major criteria are VEGF (Vascular endothelial growth factor) elevated or vascular endothelial growth factor, polyneuropathy, monoclonal plasma-proliferative disorder, sclerotic bone lesion, and Castleman disease. Minor criteria are sclerotic bone lesions, edema, papilledema, endocrine, and skin changes such as hypertrichosis, hemangioma, raynauds, white nails, clubbing, thrombocytosis, and polycythemia. Castleman disease is the term applied to the presence of angiofollicular lymph node hyperplasia. Fever and weight loss, pachymeningitis, and strokes can occur. Treatment involves multidisciplinary approach with treatment of all accompaniments, treatment of bone lesions, plasmapheresis, autologous stem cells transplant, thalidomide, lenalidomide, bortezomib, bevacizumab, an anti-VEGF antibody, etc., IgM neuropathy is less responsive to plasma exchange than IgA- or IgG-associated ones. Rituximab, interferon alpha, and thalidomide are also tried.
Role of electrophysiology
Electrophysiology is useful in confirming acquired inflammatory neuropathy from inherited ones, quantify the affected nerves, screen for variants and Electromyography pattern, and predicting prognosis.[14]
Biopsy
Biopsy of the nerves is very useful in confirming the diagnosis, quantifying the pathology, and predicting outcome. Pathological changes are distinct in AIDP, CIDP, and vasculitic neuropathy. Vasculitic neuropathy shows inflammatory infiltrate within the vessel wall and one or more signs of vascular destruction such as fibrinoid necrosis, vascular/perivascular hemorrhage, or endothelial cell disruption; Loss/fragmentation of internal elastic lamina; loss/fragmentation/separation of smooth muscle cells in media; and acute thrombosis; leukocytoclasia. Chronic lesion shows signs of healing or repair: nerve biopsy showing mononuclear inflammatory cells in the vessel wall and one or more of the following: intimal hyperplasia; fibrosis of the media; adventitial/periadventitial fibrosis; and chronic thrombosis with recanalization.
Miscellaneous immune-mediated neuropathies
Vasculitis of varying types, sarcoid, and Behcets are also secondary immune-related syndromes. Each of the subtypes has further criteria which are more specific for the subtype.
PATIENTS AND METHODS
Fifty consecutive patients seen as in patient by the authors in the last 3 years were evaluated in detail clinically. All of them underwent CSF study, electrocardiography, electrolytes, blood sugar, electrophysiological studies motor conduction velocity, sensory conduction velocity, F-wave latency of both median, ulnar common peroneal nerve, and sural nerves. HBsAG, HIV in all cases. Vasculitic workup and other workup were done based on clinical requirements. Patients were classified as AIDP based on NINCDS criteria[15] and CIDP based on American academy of neurology criteria.[116] Figure 1 shows classical histopathological features in AIDP in our case series. In AIDP, unless there are atypical features, there is no indication for nerve biopsy. Biopsy shows subperineurial edema, with inflammation both endoneurial and perivascular with active demyelination. Figure 2 shows classical features in CIDP in our case series. In CIDP, there is definitive indication for biopsy to establish the diagnosis. Patients’ nerve biopsy shows endoneural inflammation, onion bulb formation, and active remyelination. Figure 3 shows skin changes in a patient with vasculitic neuropathy.
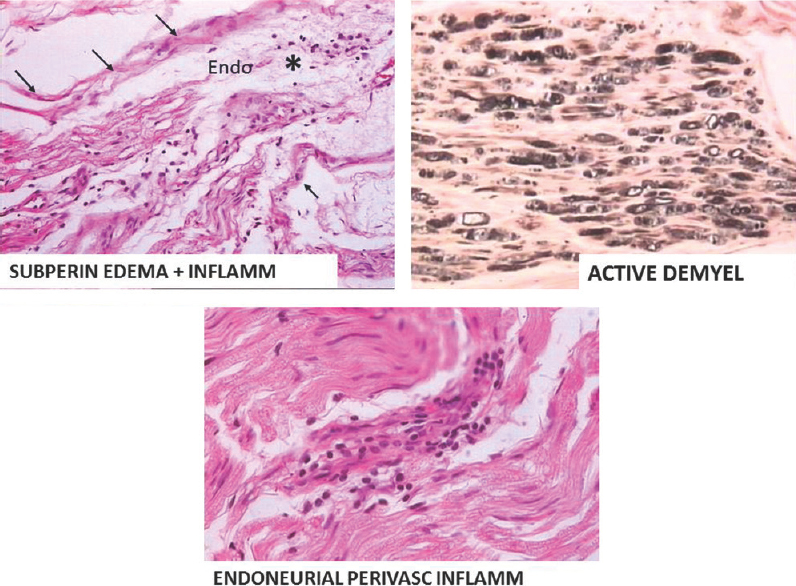
- Subperineurial edema, endoneurial and perivascular inflammation, and active demylelination

- Histopathology of chronic inflammatory demyelinating polyneuropathy showing endoneural inflammation, onion bulb formation and remylelination
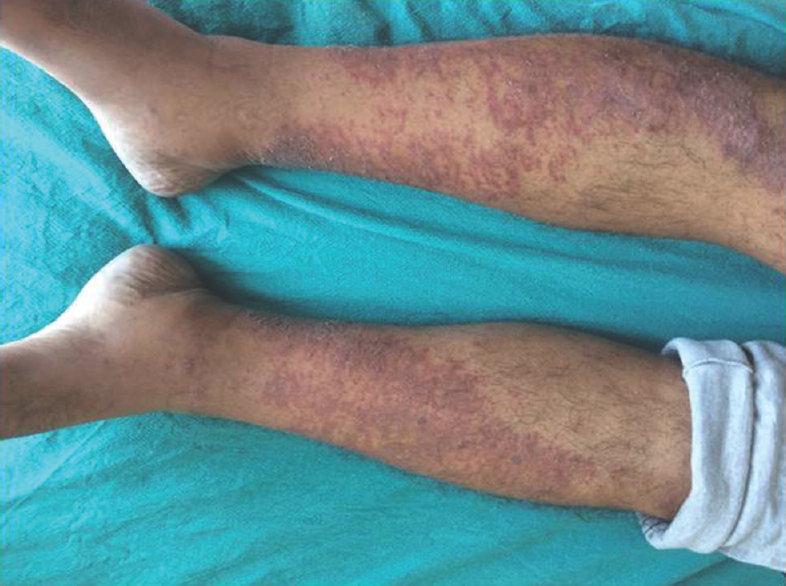
- Erythematous patch in a patient with vasculitic neuropathy
RESULTS
We had a total of sixty patients. Thirty-one were acute and 29 were chronic [Figure 4]. There were 25 male and 6 female acute cases, age range varied from 2 to 70 years. Miller fisher variant was seen in four cases. AMAN variant was noted in 1 case, paraplegic type 1 case, motor dominant type 19 cases, sensorimotor 1 case, MADSAM type 3 cases, and bifacial type 2 cases [Figures 5 and 6]. Figure 7 shows AIDP subtypes; Figure 8 shows CIDP subtypes. CIDP cases were 29 and age distribution was 27–74 years, males 19 and females 10 and vasculitic cause in 16 and HIV associated in 1 case; POEMS syndrome in 1 case presented with hypertrichosis, telangiectasia, edema, trophic changes, and osteosclerotic bone lesion. He also had M band in serum electrophoresis [Figure 9]. Juvenile diabetes and CIDP was noted in 1 case, chronic relapsing in 3 cases, and classical in 7 cases.
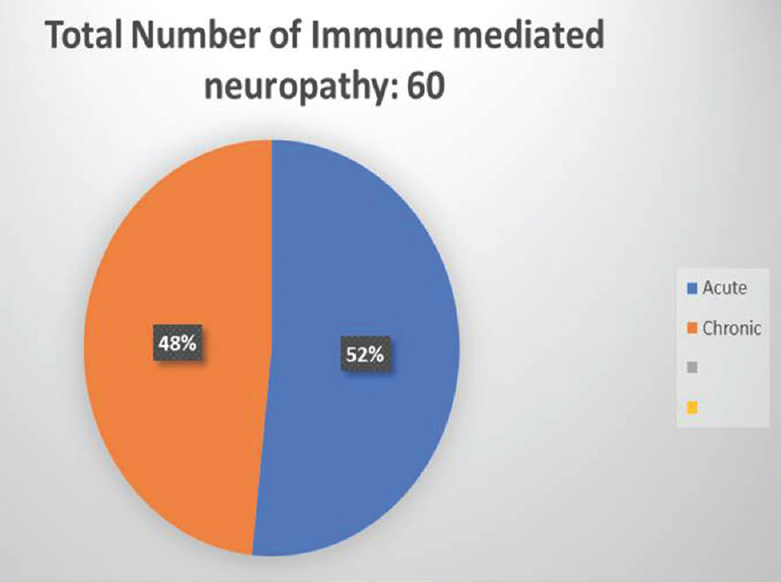
- The distribution of acute and chronic neuropathies in our series
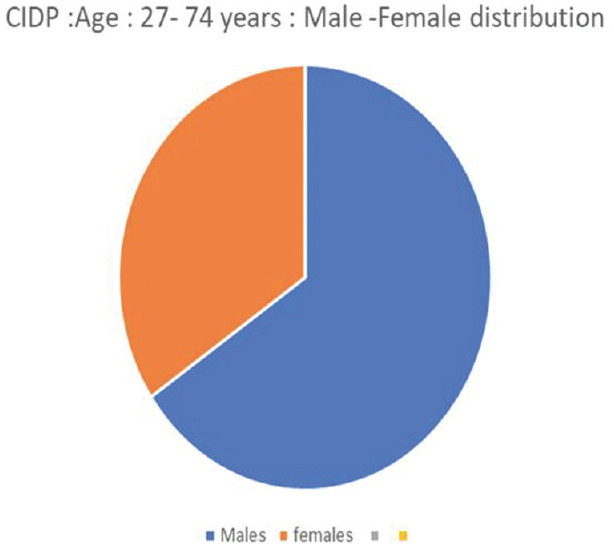
- Gender distribution in chronic inflammatory demyelinating polyneuropathy
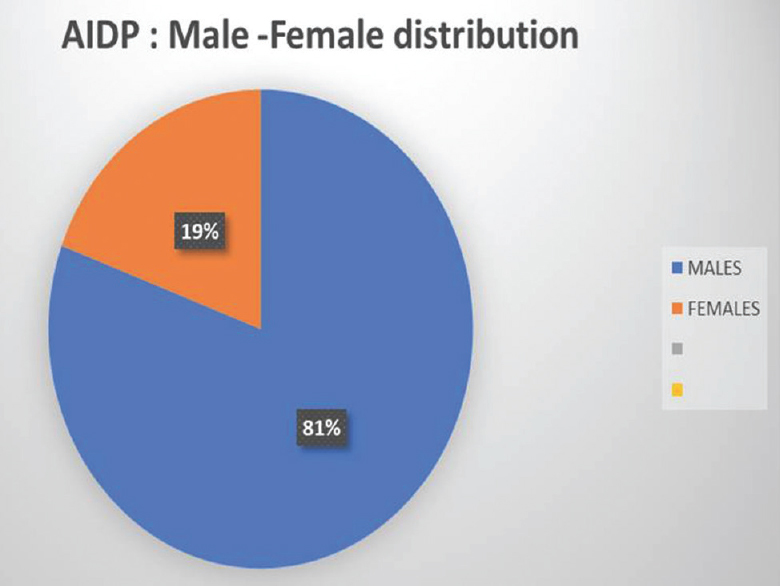
- Gender distribution in acute inflammatory demyelinating polyneuropathy patients
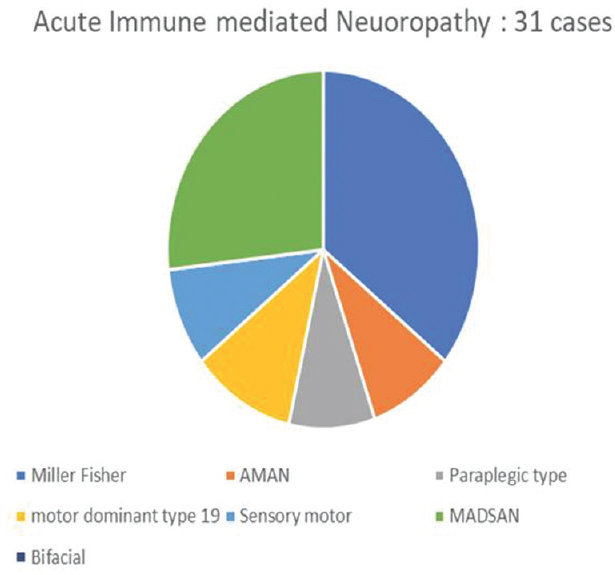
- Acute inflammatory demyelinating polyneuropathy subtype distribution
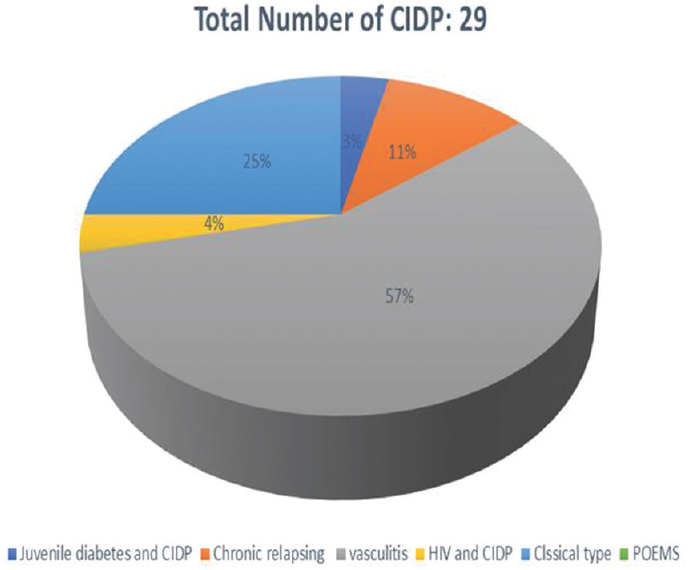
- Chronic inflammatory demyelinating polyneuropathy subtype distribution
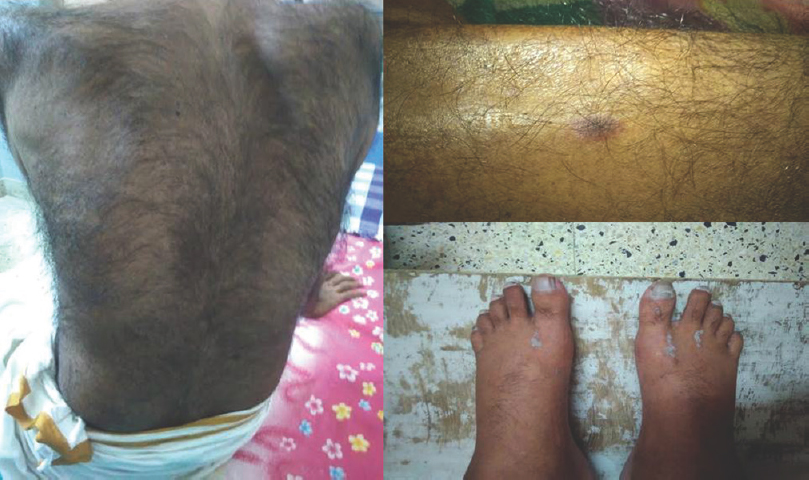
- Hypertrichosis, telangiectasia, edema legs with psoriasiform skin changes (This is typical of poems syndrome)
Treatment
All patients with AIDP received supportive treatment and plasmapheresis. Two of the patients needed intravenous immunoglobulin in view of poor response to plasmapheresis. Two patients needed ventilator care. There was no mortality in this group. Patients with CIDP were treated with steroids 1–3 mg/kg and azathioprine 2.5 mg/kg or methotrexate or cyclophosphamide 1–2 mg/kg or cyclosporine 3–7 mg/kg to start with and maintain with 1-3 mg/kg added for steroid-sparing effect. If there is no response to steroids in 6 weeks, 0.4 mg/kg/day IVIg given for 5 days monthly, low dose steroid pulses can be added. Rituximab, a CD20 monoclonal antibody, can also be tried. We used azathioprine in 23 cases and 6 patients received cyclophosphamide-based on poor treatment response.
Outcome
Our follow-up for both groups is 6 months to 3 years. There was no mortality in either group. Two patients in AIDP group needed >1 year to recover. Among CIDP patients out of 29 at the time of the last follow-up, mild sensory symptoms were reported in nine cases. Motor deficits recovered in all cases.
DISCUSSION
Immune mediated neuropathies are a large spectrum of neuropathies. They may be acute Vs Chronic, mildly disabling to serious ones, can be associated with systemic illness or occur involving peripheral nerves only. recognition of the condition in its totality is important for deciding optimum treatment, planning prognosis and predicting long term outcome.
CONCLUSION
In this 3-year follow up the study of patients seen by the authors, there was no mortality. Morbidity was very less in both groups. Therefore, it is mandatory to recognize these patients early and start treatment early so that there is very good quality of life.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- Pathogenesis and treatment of immune-mediated neuropathies. Ther Adv Neurol Disord. 2009;2:261-81.
- [Google Scholar]
- Clinical spectrum of chronic acquired demyelinating polyneuropathies. Muscle Nerve. 2001;24:311-24.
- [Google Scholar]
- Treatment of guillain-barré syndrome with mycophenolate mofetil: A pilot study. J Neurol Neurosurg Psychiatry. 2007;78:1012-3.
- [Google Scholar]
- A prospective, multicenter, randomized phase II study to evaluate the efficacy and safety of eculizumab in patients with Guillain-Barré syndrome (GBS): Protocol of Japanese eculizumab trial for GBS (JET-GBS) JMIR Res Protoc. 2016;5:e210.
- [Google Scholar]
- Assessment of current diagnostic criteria for Guillain-Barré syndrome. Ann Neurol. 1990;27(Suppl):S21-4.
- [Google Scholar]
- Clinical features, pathogenesis, and treatment of Guillain-Barré syndrome. Lancet Neurol. 2008;7:939-50.
- [Google Scholar]
- Classifications and treatment responses in chronic immune-mediated demyelinating polyneuropathy. Neurology. 2007;68:1622-9.
- [Google Scholar]
- Research criteria for diagnosis of chronic inflammatory demyelinating polyneuropathy (CIDP) Neurology. 1991;41:617-8.
- [Google Scholar]
- American Association of Electrodiagnostic Medicine. Consensus criteria for the diagnosis of multifocal motor neuropathy. Muscle Nerve. 2003;27:117-21.
- [Google Scholar]
- Electrophysiological diagnosis of guillain-barré syndrome subtype: Could a single study suffice? J Neurol Neurosurg Psychiatry. 2015;86:115-9.
- [Google Scholar]
- Diagnostic and classification criteria for the Guillain-Barré syndrome. Eur Neurol. 2001;45:133-9.
- [Google Scholar]




