
Translate this page into:
Hypercoagulability in hereditary hemorrhagic telangiectasia with epilepsy
Address for correspondence: Dr. Josef Finsterer, Postfach 20, 1180 Vienna, Austria, Europe. E-mail: fifigs1@yahoo.de
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Recent data indicate that in patients with hereditary hemorrhagic teleangiectasia (HHT), low iron levels due to inadequate replacement after hemorrhagic iron losses are associated with elevated factor-VIII plasma levels and consecutively increased risk of venous thrombo-embolism. Here, we report a patient with HHT, low iron levels, elevated factor-VIII, and recurrent venous thrombo-embolism. A 64-year-old multimorbid Serbian gipsy was diagnosed with HHT at age 62 years. He had a history of recurrent epistaxis, teleangiectasias on the lips, renal and pulmonary arterio-venous malformations, and a family history positive for HHT. He had experienced recurrent venous thrombosis (mesenteric vein thrombosis, portal venous thrombosis, deep venous thrombosis), insufficiently treated with phenprocoumon during 16 months and gastro-intestinal bleeding. Blood tests revealed sideropenia and elevated plasma levels of coagulation factor-VIII. His history was positive for diabetes, arterial hypertension, hyperlipidemia, smoking, cerebral abscess, recurrent ischemic stroke, recurrent ileus, peripheral arterial occluding disease, polyneuropathy, mild renal insufficiency, and epilepsy. Following recent findings, hypercoagulability was attributed to the sideropenia-induced elevation of coagulation factor-VIII. In conclusion, HHT may be associated with hypercoagulability due to elevated factor-VIII associated with low serum iron levels from recurrent bleeding. Iron substitution may prevent HHT patients from hypercoagulability.
Keywords
Antithrombotic agents
blood coagulation factors
risk factors
thrombophilia
thrombosis

Introduction
Hereditary hemorrhagic teleangiectasia (HHT) also known as Rendu-Osler-Weber's disease, is an autosomal-dominant vascular dysplasia, characterized by multi-focal widenings of distal arteries and a high prevalence of vascular malformations.[1] Vascular malformations include arterio-venous malformations (AVMs) in various parenchymatous organs. Cavernomas, or dural arterio-venous fistula.[2] Cerebral vascular malformations in HHT are most frequently low-grade AVMs (Spitzer–Martin grade 2). HHT may be complicated by a number of cerebral diseases, such as ischemic stroke,[3] intra-cerebral bleeding,[1] central nervous system infection,[4] cerebral abscess,[5] or epilepsy in case of a cerebral vascular malformation.[36] The risk of intra-cerebral hemorrhage is low in HHT, particularly if a pulmonary AVM is present.[2] Among those who experience intra-cerebral hemorrhage the outcome is favorable.[7] Furthermore, cerebral abscess is not unusual in HHT and develops in about 1% of the patients.[4] Epilepsy in HHT is most frequently due to the presence of cerebral vascular malformations.[8] In the absence of a cerebral vascular malformation, epilepsy is only rarely associated with HHT.[910111213] Even more rare is the association of HHT with hypercoagulability, although it has been recently shown that, sideropenia due to hemorrhagic iron losses is associated with elevated plasma levels of coagulation factor-VIII,[14] as in the following case.
Case Report
The patient is a 64-year-old (April 2012) Serbian gipsy, height 175 cm, weight 55 kg, with a history of arterial hypertension, hyperlipidemia requiring statin therapy, nicotine abuse, peripheral arterial occluding disease, and recurrent epistaxis since several years. In 2007 he experienced thrombosis of the mesenteric veins, requiring partial resection of the small intestines. In January 2009 portal venous thrombosis occurred, which is why phenprocoumon was started. In May and June 2009 he underwent osteoplastic trepanation for a cerebral, fronto-parietal abscess with a left cerebellar stroke periprocedurally. Since at least February 2010 sideropenic anemia was recognized, requiring iron substitution. Hemiglobinopathy was excluded as the cause of iron loss.[15] Shortly afterwards deep venous thrombosis of the right lower limb occurred. Since at least April 2010 he experienced recurrent cloni of the right lower leg. In April 2010, during a phenprocoumon pause for gastro-intestinal bleeding, a further ischemic stroke in the left occipital region and one day later in the left temporal area occurred. Diagnostic work-up for suspected thrombophilia in February and March 2010 revealed a markedly increased factor-VIII [Table 1]. There was no indication for severe ventricular rhythm abnormalities, pauses or atrial fibrillation or flutter. Echocardiography did not show systolic dysfunction, intracavitary thrombus formation, or noncompaction. Computed tomography (CT)-scans of the cerebrum, thorax, and abdomen revealed pulmonary and renal AVMs exclusively. In April 2010 HHT was suspected for the 1st time. In July and October 2010 an ileus of the small intestines occurred. Diabetes mellitus was first diagnosed in May 2009 and required permanent treatment since October 2011. In July 2011 polyneuropathy was diagnosed. The family history was positive for epistaxis in his father, uncle, daughter, and grandchild.
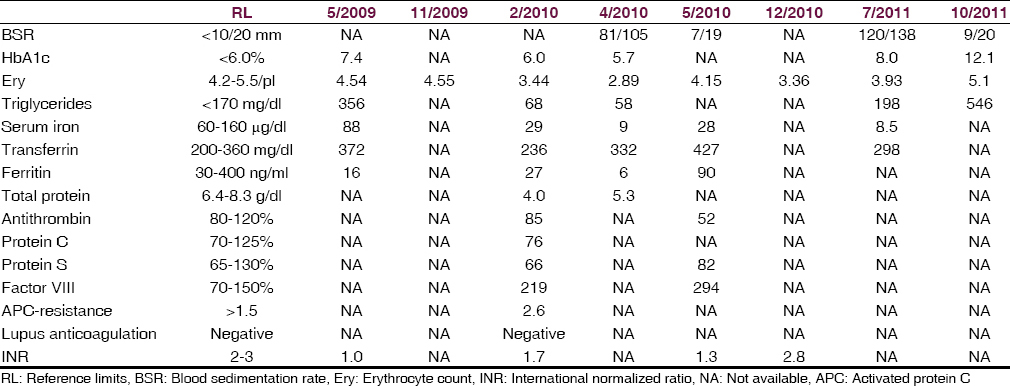
Neurological investigations at age 63 and 64 years revealed discrete hemiparesis on the right side (M5-) with distal predominance on the lower limb (M2), and an exaggerated right patella reflex but otherwise reduced tendon reflexes. Abnormal blood tests are given in Table 1. Cerebral CT in July 2011 showed old ischemic lesions in the cerebellum bilaterally, and hyperdensities of the putamina bilaterally. Cerebral magnetic resonance imaging (MRI) (December 2011) showed generalized diffuse atrophy, extensive subcortical gliosis with left-sided predominance, and multiple, spot-like lesions in the frontal white matter bilaterally. Cerebral MRI (March 2012) showed old ischemic lesions in the cerebellum bilaterally and in the left parieto-occipital region. Nerve conduction studies (December 2011) revealed reduced nerve conduction velocity of the right peroneal nerve and the median nerves bilaterally and a nonexcitable sural nerve. Polyneuropathy was diagnosed.
In the absence of documented paroxysmal activity on electroencephalogram (EEG), cloni of the right leg were interpreted as focal seizures, which is why levetiracetam was started in March 2011 and increased to a maximal dosage of 3000 mg/d. In July 2011 gabapentin was added and increased to a maximal dosage of 1800 mg/d. Under this regimen focal seizures, which predominantly occurred during the night, stopped for about 2 months, but recurred in November 2011, which is why gabapentin was increased to 2400 mg/d. Despite this increase, seizures persisted, and levetiracetam was intended to be replaced by carbamazepine. Unfortunately, he did not tolerate carbamazepine and developed tremor of the upper limbs and unsteadiness. In March 2012 EEG, for the 1st time, showed spikes and spike-wave complexes in the left centro-parieto-temporal regions, which is why carbamazepine was replaced by lacosamide. At his last visit he was on a medication with levetirazetam (2000 mg/d), lacosamide (100 mg), metformin (2500 mg/d), glimepirid (1 mg/d), and iron-sulfate, but nonetheless serum iron levels were reduced [Table 1].
Discussion
The presented patient is interesting for the co-occurrence of HHT and recurrent venous thrombosis due to increased factor-VIII hypercoagulability. HHT was diagnosed according to the Curacao criteria.[7] The patient had a history of recurrent epistaxis, teleangiectasias on the lips, AVMs in the kidneys and lungs, and a positive family history for the disease. Hypercoagulability was diagnosed on the history with recurrent, multi-focal venous thromboses and blood coagulation tests. Contrary to HHT, hypercoagulability was absent in other family members.
Though venous thrombosis has been occasionally reported in HHT,[1516] the co-occurrence of HHT and hypercoagulability due to an elevation of factor-VIII, as in the presented case, is rare. To which degree both abnormalities influenced each other and contributed to the clinical presentation, remains speculative. Since other plausible causes of venous thrombosis were absent in the presented patient, recurrent thromboses were attributed to the elevation of factor-VIII.[14] That thrombosis occurred despite oral anticoagulation is unusual but can be explained by the recurrent sub-therapeutic international normalized ratio-values, by impaired intestinal resorption of phenprocoumon, or generally by too low reference values. Whether patients with HHT, venous thrombosis and atherosclerosis should generally receive oral anticoagulation, antithrombotic agents alone or in combination or none of them, is questionable, particularly in the light of their increased bleeding risk due to their vascular dysplasia. However, those with hypercoagulability might profit from oral anticoagulation and those with atherosclerosis from antithrombotic treatment as long as their iron levels remain normal, and no acute or chronic bleedings occur. Recurrent ischemic strokes in the presented patient presumably occurred because of poor risk factor control or abandoned antithrombotic treatment. Following the experience with this patient it has to be guaranteed that patients with HHT have normal serum iron levels to prevent hypercoagulability and to provide antithrombotic treatment if indicated.
This case shows that HHT may be associated with elevated factor-VIII-induced hypercoagulability due to low serum iron levels from recurrent bleeding and that epilepsy may be a feature of HHT even in the absence of a cerebral AVM.
Source of Support: Nil.
Conflict of Interest: None declared.
References
- Bleeding risk of cerebrovascular malformations in hereditary hemorrhagic telangiectasia. J Neurosurg. 2000;92:779-84.
- [Google Scholar]
- Cerebrovascular manifestations in 321 cases of hereditary hemorrhagic telangiectasia. Stroke. 2001;32:877-82.
- [Google Scholar]
- Pulmonary arteriovenous malformations: Cerebral ischemia and neurologic manifestations. . 2000;55:959-64.
- [Google Scholar]
- Central nervous system infections associated with hereditary hemorrhagic telangiectasia. Am J Med. 1984;77:86-92.
- [Google Scholar]
- Familial brain abscess as a complication of hereditary hemorrhagic telangiectasia. Orv Hetil. 2001;142:2309-12.
- [Google Scholar]
- Growth and regression of arteriovenous malformations in a patient with hereditary hemorrhagic telangiectasia. Case report. J Neurosurg. 2007;106:470-7.
- [Google Scholar]
- The prevalence and manifestations of hereditary hemorrhagic telangiectasia in the Afro-Caribbean population of the Netherlands Antilles: A family screening. Am J Med Genet A. 2003;116A:324-8.
- [Google Scholar]
- Unsuspected mesenteric vein thrombosis in a patient with a hereditary bleeding disorder. Blood Coagul Fibrinolysis. 2003;14:599-600.
- [Google Scholar]
- Vascular malformations of the brain in hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber disease) Surg Neurol. 1994;41:374-80.
- [Google Scholar]
- Article in French Rendu-Osler disease and epilepsy. Rev Neurol (Paris). 2000;156:794-5.
- [Google Scholar]
- Rendu-Osler-Weber disease and cerebral arteriovenous malformation. Neurologia. 1988;3:83-4.
- [Google Scholar]
- Progressive somnolence and confusion in a patient with hereditary hemorrhagic telangiectasias. Tenn Med. 1996;89:417-8.
- [Google Scholar]
- Cleidocranial dysostosis, hereditary haemorrhagic telangiectasia and epilepsy: A rare association. Neuroradiology. 1977;12:233-6.
- [Google Scholar]
- Autonomic seizures in Rendu-Osler-Weber disease. Neurol Neurochir Pol. 1967;1:409-11.
- [Google Scholar]
- Low serum iron levels are associated with elevated plasma levels of coagulation factor VIII and pulmonary emboli/deep venous thromboses in replicate cohorts of patients with hereditary haemorrhagic telangiectasia. Thora×. 2012;67:328-33.
- [Google Scholar]




