
Translate this page into:
Epstein-Barr virus induced hemophagocytic lymphohistiocytosis in X-linked lymphoproliferative disease
Address for correspondence: Dr. Rosario Maria Riel-Romero, Department of Pediatrics and Neurology, Louisiana State University Health Sciences Center, 1501 Kings Highway, Shreveport, LA, 71130, USA. E-mail: rrielr@lsuhsc.edu
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
X-linked lymphoproliferative disease (XLP) is a rare, often fatal genetic disorder characterized by extreme vulnerability to Epstein-Barr virus (EBV). EBV-induced hemophagocytic lymphohistiocytosis (HLH) is a known presentation in XLP. In EBV-induced HLH in XLP, the brain imaging findings in the acute phase include a non specific pattern. In this report, we highlight the magnetic resonance imaging and magnetic resonance spectroscopy findings in a child with EBV induced HLH in XLP.
Keywords
Epstein-Barr virus
hemophagocytic lymphohistiocytosis
X-linked lymphoproliferative disease

Introduction
X-linked lymphoproliferative disease (XLP) is a rare, often fatal genetic disorder characterized by extreme vulnerability to Epstein-Barr virus (EBV) infections. The clinical manifestations of XLP are variable and characterized by severe immune dysregulation. The phenotypic expression includes fulminant infectious mononucleosis, hemophagocytic lymphohistiocytosis (HLH), lymphoma and dysgammaglobulinemia, often triggered by EBV infection.[123] In EBV-induced HLH in XLP, the brain imaging findings in the acute phase include a non specific inflammatory pattern and demyelination. In this report, we highlight the magnetic resonance imaging (MRI) and magnetic resonance spectroscopy (MRS) findings in a child with EBV induced HLH in XLP. The clinical and laboratory details of this patient were previously reported by our colleagues (Kaza et al).[4]
Case Report
The patient, a 4.5-year-old male without a significant past medical history, presented with fever for two weeks and subsequently developed a progressive downhill course characterized by lymphadenopathy and hepatosplenomegaly.[4] Family history (death of a cousin from lymphoproliferative disease) provided clue for the possibility of XLP. Cerebrospinal fluid analysis, lymph node and bone marrow biopsies demonstrated EBV infection and HLH was confirmed as per the Histiocyte Society HLH protocol.[45] XLP was confirmed by reduced expression of SLAM (signaling lymphocyte-activation molecule)-associated protein (SAP) and genetic sequencing confirmed mutation in the SH2D1A gene.[4] Despite aggressive treatment with multiple agents, he deteriorated and died.[4] On initial presentation, the brain MRI demonstrated widespread signal changes in the cerebral white matter, cerebellum and in the basal ganglia [Figure 1a–c and Figure 2a–f]. Lesions in the right temporal region, left basal ganglia and cerebellum [Figure 2a–c] showed marked enhancement with contrast (Gadolinium-DTPA) administration [Figure 2d–f]. There was also hemorrhage in the right temporal lesion [Figure 2b]. Repeat imaging a month later showed increased size of the lesions [Figure 1d–f]. MRS during the acute phase showed the presence of lactate and lipid peaks indicating anaerobic metabolism and necrosis [Figure 3]. Additionally, the decreased N-acetyl aspartate (NAA) peak suggested significant neuronal loss and also mild elevation of choline (Cho) representing increased neuronal membrane turnover. The presence of widespread lesions on MRI and the MR spectroscopy findings are consistent with inflammatory necrotic damage.
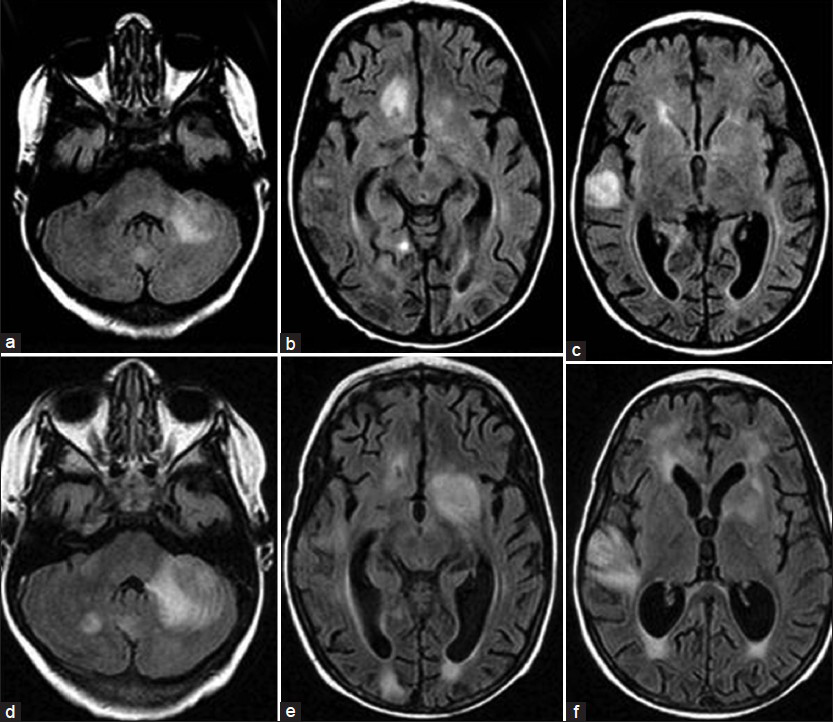
- Magnetic Resonance. FLAIR sequence. Transverse slices (a, b, c) at initial presentation and (d, e, f) taken one month later. Note the increased size of lesions involving mainly the white matter, but extending to the cortex. The most remarkable lesions are located in the left middle cerebellar peduncle, left cerebellar hemisphere right temporal polar region and left basal ganglia
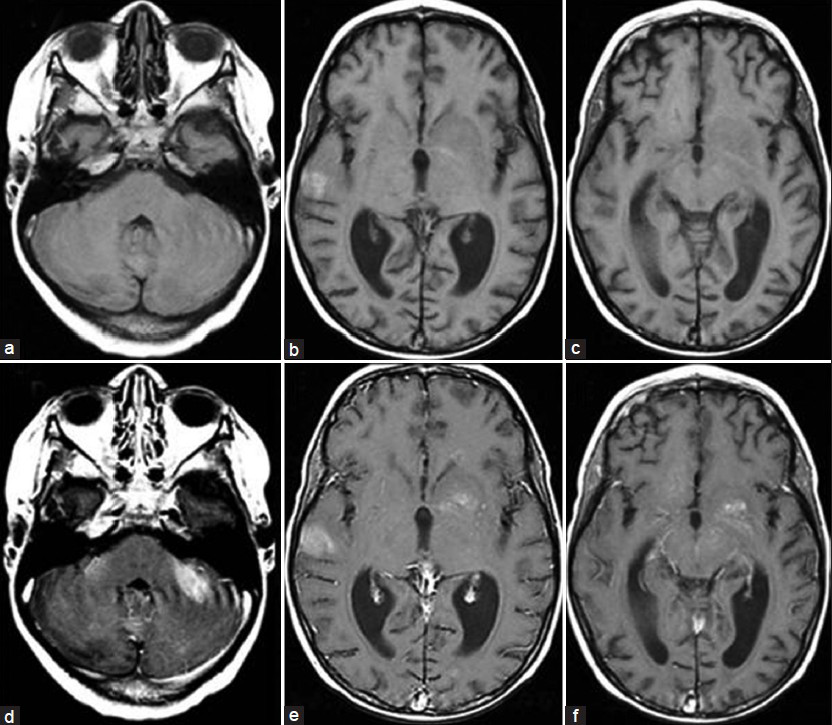
- Magnetic resonance. T1W sequence at initial presentation. Transverse plane (a, b, c) before contrast (d, e, f) after contrast (Gd-DTPA) administration. Marked enhancement of the cerebellar, right temporal and left basal ganglia lesions were noted. Hyperintensity on the right temporal lesion seen before contrast indicate the presence of blood
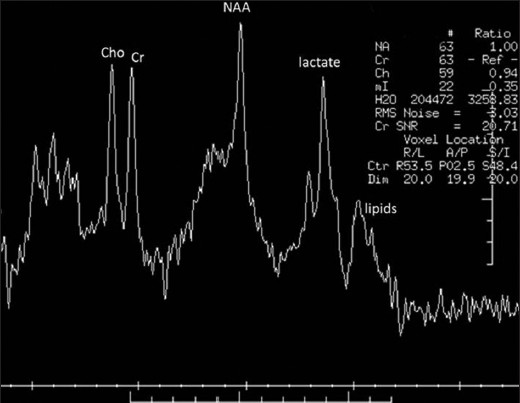
- Magnetic resonance spectroscopy. Presence of lipid and lactate peaks indicates anaerobic metabolism and necrosis. Decreased N-acetyl aspartate peak suggests neuronal loss. Mild increase in choline suggests increased neuronal membrane turnover
Discussion
X-linked lymphoproliferative disease (XLP) was first described in 1975 by Purtilo in the Duncan family.[6] Overall, XLP affects approximately 1 in one million males. The survival of XLP patients is very poor despite aggressive treatment, and the majority of patients die in childhood.[3] The clinical syndrome of XLP occurs due to 2 different genetic defects - SH2D1A (XLP1, the most common defect) and the BIRC/XIAP gene (XLP2).[3]
Hemophagocytic lymphohistiocytosis (HLH) is one of the known presentations of XLP. HLH is a clinical syndrome of hyperinflammation as a result of deficient down-regulation of the immune response.[7] HLH develops either subsequent to genetic mutations (familial form) or in association with infection, malignancy, autoinflammatory or metabolic conditions (acquired form).[7] Regardless of the type of HLH, central nervous system (CNS) involvement has been reported in 10-73% of patients, either at initial presentation or during the course of the disease.[8] The presence of CNS involvement at diagnosis is a poor prognostic sign, and requires prompt management.[9] Neurological presentations are variable and include irritability, seizures, ataxia, cranial nerve palsies, meningism, signs of increased intracranial pressure, and altered consciousness.[8] The neuroradiological findings are not distinguishable between the familial and acquired forms of HLH.[9] Rego et al. described three different patterns of parenchymal involvement: Diffuse, focal and mixed diffuse/focal involvements.[9] The neuroradiological findings in HLH are not sufficiently specific to achieve the diagnosis. The list of differential diagnoses includes brain abscess, metastasis, multiple sclerosis, lymphoma, acute disseminating encephalomyelitis or other meningoencephalitis, chemotherapy-induced leucoencephalopathy, malignant glioma, acute cerebral infarction, and child abuse.[8]
Only few case reports describe the imaging findings of EBV induced HLH in XLP.[19] Milshcher et al. described widespread, non-enhancing signal abnormalities predominantly seen in the frontal white matter and cerebellar peduncles.[1] Rego et al. described diffuse/focal cortical and basal ganglia lesions.[9] The metabolic changes observed in MRS by Rego et al. in acute HLH were indicative of active demyelination/inflammation with decreased NAA representing axonal dysfunction, increase in choline correlating with increased neuronal membrane turnover and presence of lactate reflecting anaerobic metabolism.[9] During follow-up new diffuse white matter and cortical lesions, increased brain atrophy were noted. Also follow-up MRS revealed decreased NAA indicating neuronal loss.[9]
As EBV induced HLH in XLP is often fatal, early prompt recognition and aggressive management is mandatory to reduce morbidity and mortality. This report emphasizes the spectrum of neuroimaging abnormalities associated with this rare entity. Even though not specific, identification of neuroimaging abnormalities in HLH is a poor prognostic sign. Hence in the presence of neurological manifestations, appropriate neuroimaging is recommended in the management of HLH.
Source of Support: Nil.
Conflict of Interest: None declared.
References
- Epstein-Barr virus-induced hemophagocytic lymphohistiocytosis and X-linked lymphoproliferative disease: A mimicker of sepsis in the pediatric intensive care unit. Pediatrics. 2007;119:e1212-8.
- [Google Scholar]
- X-linked lymphoproliferative disease due to SAP/SH2D1A deficiency: A multicenter study on the manifestations, management and outcome of the disease. Blood. 2011;117:53-62.
- [Google Scholar]
- Clinical and genetic features of 5 Chinese patients with X-linked lymphoproliferative syndrome. Scand J Immunol. 2013;78:463-7.
- [Google Scholar]
- A boy with fever, lymphadenopathy, hepatosplenomegaly, and lymphocytosis. Allergy Asthma Proc. 2008;29:216-20.
- [Google Scholar]
- HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48:124-31.
- [Google Scholar]
- X-linked recessive progressive combined variable immunodeficiency (Duncan's disease) Lancet. 1975;1:935-40.
- [Google Scholar]
- A spectrum of neuroradiological findings in children with haemophagocytic lymphohistiocytosis. Pediatr Radiol. 2007;37:1110-7.
- [Google Scholar]
- Neuroradiologic findings and follow-up with magnetic resonance imaging of the genetic forms of haemophagocytic lymphohistiocytosis with CNS involvement. Pediatr Blood Cancer. 2012;58:810-4.
- [Google Scholar]




