
Translate this page into:
Cranial and Spinal Malignant Peripheral Nerve Sheath Tumor: A Pathological Enigma
 , Sumitra Sivakoti3, Vani Santosh4, Sampath Somanna2
, Sumitra Sivakoti3, Vani Santosh4, Sampath Somanna2
Manish Beniwal, MCh Department of Neurosurgery, National Institute of Mental Health and Neurosciences Bengaluru 560029, Karnataka India beniwal.m@gmail.com
This article was originally published by Thieme Medical and Scientific Publishers Pvt. Ltd. and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Abstract
Objective Malignant peripheral nerve sheath tumor (MPNST) arises from nerve sheaths, mostly seen in peripheral nerves but rare in craniospinal nerves. The information available in the literature to build up treatment strategy and improve clinical outcomes is scarce. We are reviewing cases from our institute, with emphasis on radiological features for early differentiation from its benign variants.
Methods We analyzed pathologically diagnosed cases retrospectively from January 2007 to December 2018 at our institute. Clinicoradiological details and treatment parameters were collected from medical records for evaluation. Each case was contacted telephonically for final clinical follow-up at the time of writing the manuscript.
Results A total of seven cases of MPNST were diagnosed in the last 10 years. It included four intracranial and three spinal cases. The mean age for the cohort was 34.3 years, with five females. We could achieve gross total resection (GTR) and subtotal resection in four (57.1%) and two (28.6%) cases, respectively. We could achieve an overall survival of 57.1% in the average follow-up of 28.2 months (range: 8–84 months).
Conclusion MPNST is a rare tumor with a bad prognosis. Radical surgical resection is the mainstay of the treatment, but it is not always possible to achieve it because of the inaccessible location and large size of lesions. Preoperative diagnosis is challenging; however, few radiological findings may give a clue toward it. As a disease entity overall, it has a poor outcome with a high rate of fatality.
Keywords
MPNST
intracranial
intraspinal
cranial nerve MPNST
radiology
treatment

Introduction
A malignant peripheral nerve sheath tumor (MPNST) is a malignant tumor arising from nerve sheath cells or cells differentiating toward it. While accounting for 5 to 10% of all soft tissue sarcomas,1 these rare tumors have an incidence as low as 0.001% in the general population.2 The incidence of central nervous system (CNS) MPNST is even more sporadic with aggressive behavior.3 4 However, the literature's diversity and sheer volume make it impossible to establish fundamental guidelines for diagnosis both radiologically5 and pathologically.6
These tumors have a robust association with neurofibromatosis 1 (NF1), with 4.6 to 10% of cases likely to develop MPNST.3 They arise within a normal peripheral nerve or from pre-existing nerve sheath tumors, usually intraneural or plexiform neurofibromas. The malignant transformation from a schwannoma is a much rarer phenomenon.7 The association between therapeutic irradiation and the development of MPNSTs is also well known.8
The standard of treatment for MPNST is total surgical resection,9 10 but not always possible due to the invasive nature of these tumors and critical locations.11 Radiotherapy and chemotherapy are used as adjuvant treatments for these tumors, but these are relative chemo-resistant tumors.12
Even with aggressive treatment regimens, the prognosis for intracranial or spinal MPNSTs remains poor, even worse than for MPNSTs in all other body sites.9 10
We present our experience of seven cases diagnosed at our center in various locations along the neuroaxis. Each case is unique in its clinical and radiological presentation, highlighting the importance of a high index of suspicion for diagnosing these cases.
Methods
Our institute is a dedicated neurosciences institute in the country's southern part. We performed a retrospective review of cases diagnosed pathologically as MPNST involving the craniospinal region. Clinico-radiological details and treatment parameters, along with follow-up details, were collected from the medical record section. The pathology of all the cases was confirmed after re-evaluation by the senior pathologist of the department. Each patient was contacted telephonically for final clinical follow-up at the time of writing the manuscript.
Results
Case 1
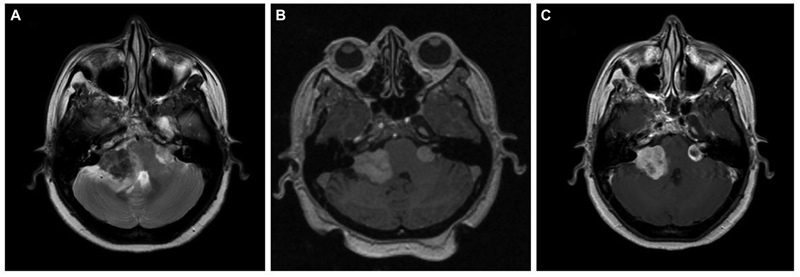
A 51-year-old female, case of NF2 with bilateral vestibular schwannomas, presented with right seventh nerve paresis and ataxic gait for 2 months. She had profound sensory neural type hearing loss (SNHL) on the right side and minimal SNHL on the left side. On magnetic resonance imaging (MRI) evaluation, right vestibular schwannoma was more significant than left. The lesion on the right side was ill-defined, hypointense on T2-weighted (T2W), isointense on T1-weighted (T1W), and heterogeneously enhancing on contrast images. The left-side tumor was small, well-defined, and was hyperintense on T2 (Fig. 1). She underwent Gamma Knife radiosurgery (GKRS) for the left-side tumor as the size was amenable, and the hearing was preserved. She received 12 Gray (Gy) for a volume of 1.3cc at 50% isodose. During the 6-month follow-up, she worsened with paraesthesia and atypical trigeminal neuralgic pain on the right side of the face. Microsurgery was performed with subtotal resection (STR), as the lesion was adherent to the brain stem. Histopathology revealed a nerve sheath tumor comprising plump spindle-shaped cells arranged in hypercellular interlacing fascicles with areas of necrosis, marked nuclear atypia, and high mitosis, indicating the malignant nature of the nerve sheath tumor. A diagnosis of MPNST was made on histopathology. She took adjuvant radiotherapy for residual right-side tumor, and at the last follow-up of 11 months after surgery, tumor size remained the same. As the left-side tumor remained the same size after GKRS and MRI characteristics were different from the right-side tumor, the left-side lesion was kept under close surveillance.
-
Fig. 1 Brain magnetic resonance imaging of a known case of neurofibromatosis-2. (A) Axial T2-weighted image showing bilateral cerebellopontine angle lesion with a typical “ice-cream cone” appearance. The right-side tumor is mainly hypointense with loss of planes with an adjacent brain stem and cerebellum with hyperintensity in the parenchyma suggestive of edema. The left side tumor is small with clear cerebrospinal fluid space around the lesion, suggesting an extra-axial tumor. (B) Axial T1-weighted image showing bilateral lesions that are predominantly isointense. (C) Contrast-enhanced post-Gamma Knife radiosurgery axial image (at 6-month follow-up) showing central hypointensity on left side tumor, suggestive of central necrosis post-treatment. Right side tumor showing heterogeneous contrast enhancement.
Fig. 1 Brain magnetic resonance imaging of a known case of neurofibromatosis-2. (A) Axial T2-weighted image showing bilateral cerebellopontine angle lesion with a typical “ice-cream cone” appearance. The right-side tumor is mainly hypointense with loss of planes with an adjacent brain stem and cerebellum with hyperintensity in the parenchyma suggestive of edema. The left side tumor is small with clear cerebrospinal fluid space around the lesion, suggesting an extra-axial tumor. (B) Axial T1-weighted image showing bilateral lesions that are predominantly isointense. (C) Contrast-enhanced post-Gamma Knife radiosurgery axial image (at 6-month follow-up) showing central hypointensity on left side tumor, suggestive of central necrosis post-treatment. Right side tumor showing heterogeneous contrast enhancement.
Case 2
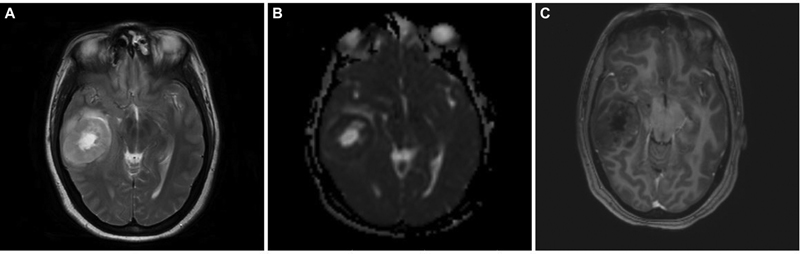
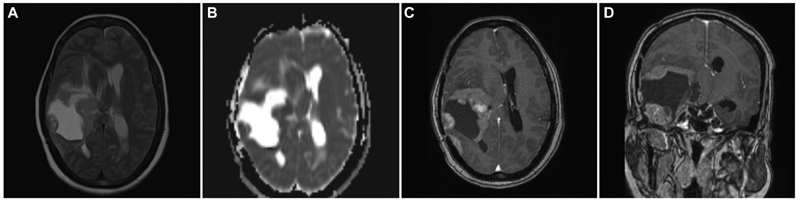
A 34-year-old female, who had headache and vomiting for 1 month, presented with an MRI brain showing an ill-defined right posterior temporal parenchymal lesion, isohyperintense on T2W with patchy enhancement on contrast. There was diffusion restriction with MR spectroscopy showing high choline:creatine ratio suggestive of a high-grade lesion pathologically (Fig. 2). She underwent gross total resection (GTR) of the lesion, followed by adjuvant radiotherapy and chemotherapy based on preoperative MRIs and postoperative histopathological diagnosis of glioblastoma. At 2 years, she developed recurrence of symptoms of raised intracranial pressure with a similar intensity lesion on MRI at the previous resection site, but with more hypointensities centrally suggesting necrotic cavity with the solid peripheral area (Fig. 3). She underwent re-exploration with GTR again. The histopathology of the second specimen is composed of elongated cells with oval vesicular nuclei arranged in fascicles extending along the Virchow-Robin spaces into the cortex. Tumor cells expressed patchy S-100 staining and were negative for glial fibrillary acidic protein suggestive of MPNST. When senior pathologists reviewed pathology slides of previous surgery in the department, it showed features of MPNST were present even in the first specimen; hence, diagnosis was revised to recurrent MPNST instead of glioblastoma. Soft tissue sarcomas contain areas of heterogeneity, which explains the difficulty in diagnosis sometimes.13 The tumor exhibited focal areas of incipient necrosis, brisk mitosis, and p53 positivity, indicating a higher grade nature of the neoplasm. She received another course of adjuvant radiotherapy for 50 days. After 2 more years, she developed raised intracranial pressure symptoms again, and on imaging, a highly infiltrative neoplasm was noted. Surgery was not performed given the poor general condition with the Karnofsky performance score of 50. She succumbed to death in 6 months, after 4 years of initial diagnosis.
-
Fig. 2 Brain magnetic resonance imaging of right temporal intraparenchymal malignant peripheral nerve sheath tumor. (A) Axial T2-weighted image showing right temporal heterogeneously hyperintense well-defined tumor with a central bright hyperintense area with perilesional edema. (B) Axial apparent diffusion coefficient (ADC) image showing lower ADC values with centrally facilitated diffusion in the lesion (equally hyperintense to ventricular cerebrospinal fluid). (C) Axial contrast-enhanced image showing centrally hypointense lesion with minimal contrast enhancement.
Fig. 2 Brain magnetic resonance imaging of right temporal intraparenchymal malignant peripheral nerve sheath tumor. (A) Axial T2-weighted image showing right temporal heterogeneously hyperintense well-defined tumor with a central bright hyperintense area with perilesional edema. (B) Axial apparent diffusion coefficient (ADC) image showing lower ADC values with centrally facilitated diffusion in the lesion (equally hyperintense to ventricular cerebrospinal fluid). (C) Axial contrast-enhanced image showing centrally hypointense lesion with minimal contrast enhancement.
-
Fig. 3 Brain magnetic resonance imaging of recurrent temporal intraparenchymal malignant peripheral nerve sheath tumor. (A) Axial T2-weighted image showing hyperintense lesion with peripheral solid nodular tumor with surrounding edema and mass effect. (B) Axial apparent diffusion coefficient images showing the peripherally isointense nodular recurrent tumor. (C) Contrast-enhanced axial image showing heterogeneous contrast enhancement of the solid tumor. (D) Contrast-enhanced coronal image showing recurrent enhancing tumor at the temporal base and the periphery of the previous surgical cavity.
Fig. 3 Brain magnetic resonance imaging of recurrent temporal intraparenchymal malignant peripheral nerve sheath tumor. (A) Axial T2-weighted image showing hyperintense lesion with peripheral solid nodular tumor with surrounding edema and mass effect. (B) Axial apparent diffusion coefficient images showing the peripherally isointense nodular recurrent tumor. (C) Contrast-enhanced axial image showing heterogeneous contrast enhancement of the solid tumor. (D) Contrast-enhanced coronal image showing recurrent enhancing tumor at the temporal base and the periphery of the previous surgical cavity.
Case 3
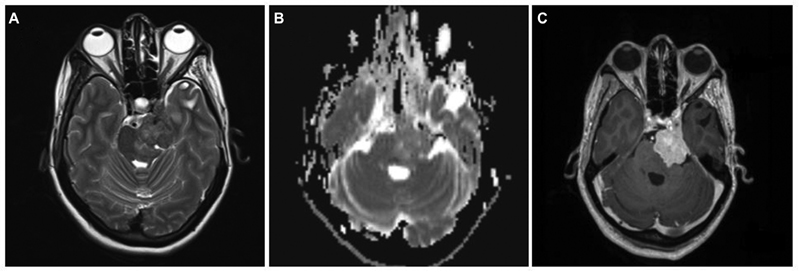
A case of a 39-year-old female who underwent subtotal excision of trigeminal schwannoma was referred to our center for GKRS for a residual lesion in the cavernous sinus. Preoperatively, she presented with complaints of facial paraesthesia and motor weakness of the Vth nerve. As the remaining lesion was small, initially, she was kept under observation. At 2 years follow-up, as the size of the residual lesion increased, she underwent GKRS. During the follow-up period, the lesion did not respond to GKRS, and the patient developed complete ophthalmoplegia with complete Vth nerve palsy. At 4 years post-GKRS, the lesion grew significantly to cause brain stem compression. On MRI, lesion was irregular, ill-defined along the Vth nerve near petrous apex extending anteriorly into the cavernous sinus region. The lesion was hypo to isointense on T2W and completely isointense on T1W with irregular enhancement on contrast (Fig. 4). The second surgery was considered given increasing tumor size and worsening neurological deficits post-GKRS. She underwent GTR of the lesion, including the cavernous part, as she had fixed preoperative neurologic deficits. The histopathological specimen after the second surgery showed neoplastic cells exhibiting nuclear atypia with prominent nucleoli and frequent mitosis. Immunohistochemically, tumor cells exhibited patchy S-100 positivity and loss of H3Me3 expression suggestive of MPNST (Fig. 5). Although the second histopathology report is of MPNST after GKRS, as we could not review slides of the first surgery, it was not possible to label it as malignant degeneration secondary to radiation. She received 54 Gy of radiotherapy over 30 fractions after the second surgery. At 8 months' follow-up, she is doing well without any new deficits and recurrence.
-
Fig. 4 Brain magnetic resonance imaging of left Vth nerve post-Gamma Knife radiosurgery recurrent tumor. (A) T2-weighted axial image showing a hyperintense lesion in cavernous sinus with posterior fossa extension with loss of arachnoid planes and brain stem edema. (B) Apparent diffusion coefficient (ADC) axial image showing mainly isointense lesion on ADC. (C) Contrast-enhanced axial image showing bright heterogeneous contrast enhancement.
Fig. 4 Brain magnetic resonance imaging of left Vth nerve post-Gamma Knife radiosurgery recurrent tumor. (A) T2-weighted axial image showing a hyperintense lesion in cavernous sinus with posterior fossa extension with loss of arachnoid planes and brain stem edema. (B) Apparent diffusion coefficient (ADC) axial image showing mainly isointense lesion on ADC. (C) Contrast-enhanced axial image showing bright heterogeneous contrast enhancement.
-
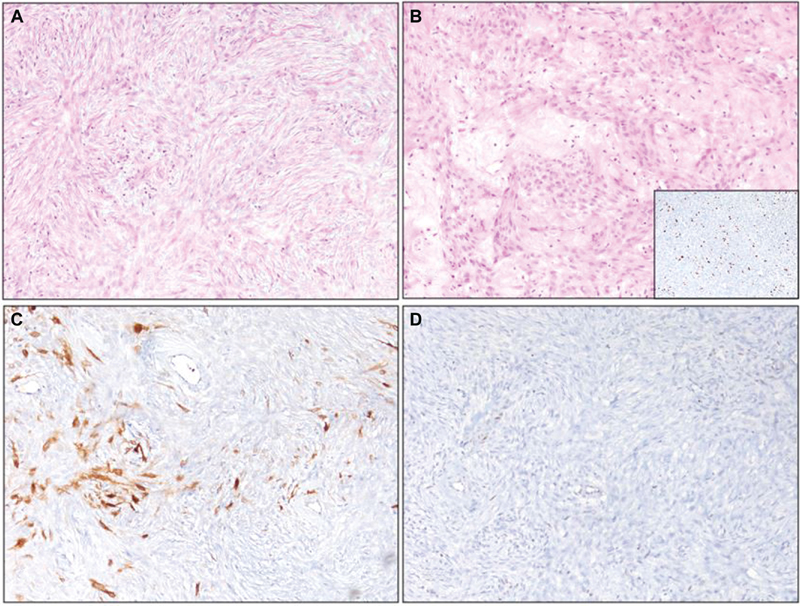
Fig. 5 Histopathology of malignant peripheral nerve sheath tumor (images magnified at 100X). (A) Neoplastic spindle cells arranged in a fasciculated pattern. (B) Numerous thickened blood vessels in the stroma with a myxoid change. Insert shows cells exhibited high MiB-1 labeling. (C) The tumor cells exhibited patchy S-100 staining. (D) Loss of H3k27me3 in neoplastic cells and retained staining in blood vessels.
Fig. 5 Histopathology of malignant peripheral nerve sheath tumor (images magnified at 100X). (A) Neoplastic spindle cells arranged in a fasciculated pattern. (B) Numerous thickened blood vessels in the stroma with a myxoid change. Insert shows cells exhibited high MiB-1 labeling. (C) The tumor cells exhibited patchy S-100 staining. (D) Loss of H3k27me3 in neoplastic cells and retained staining in blood vessels.
Case 4
An 18-year-old male presented with decreased hearing and chewing difficulty on the left side for 6 months, with a deviation of the angle of the mouth to the right. Preoperatively, he had weakness of muscles of mastication on the left side with numbness involving all three divisions of the Vth nerve. There was a grade IV House-Brackman facial palsy and profound hearing loss on the left side. On MRI, there was a lesion along the Vth nerve in a Meckel's cave area at the petrous apex with a small posterior fossa component extending up to the jugular bulb and extending extracranially through it. The lesion was isohypointense on T1W and hyperintense on T2W with diffusion restriction and heterogeneous enhancement (Sami's type D). He underwent Kawase's approach and STR of the lesion of the middle cranial fossa component. Histopathologically, it proved to be MPNST. Postoperatively, he received 59.4Gy / 33# radiotherapy. Post radiation after 6 months, he developed left side frozen eyeball with complete ptosis and hoarseness of voice. MRI showed recurrence of the lesion extending into the cavernous sinus region and with a significant posterior fossa component. He underwent excision of the intracranial segment by re-exploration of the previous Kawase approach followed by separate retromastoid craniotomy for the posterior fossa component after 15 days. Histopathological examination revealed a high-grade malignant nerve sheath tumor comprised of pleomorphic spindle cells arranged in storiform patterns and fascicles with brisk mitosis and apoptosis. He is currently on adjuvant radiotherapy for residual extracranial lesion, and presently he is doing well with the persistence of preoperative neurologic deficits.
Case 5
A 51-year-old male presented with a history of gradually progressive weakness and paraesthesia of lower limbs, and urinary hesitancy, and incomplete voiding for 2 months. He underwent a contrast-enhanced MRI spine, which showed well-defined extradural mass lesion extending from C7 (cervical 7) to D3 (dorsal 3) level located dorsally. The lesion was hypointense on T1W and T2W with homogenous enhancement on contrast (Fig. 6) On computed tomographic spine images, there was bony destruction of the D1 and D2 spinous process. He underwent GTR of this extradural intraspinal lesion, after which his lower limb power improved significantly. The histopathological examination confirmed spindle cell tumor of nerve sheath origin with variable patchy S-100 protein expression, and mitotic activity is ∼4 to 5 in 10/high-power field (HPF), suggestive of MPNST grade II. Unfortunately, we do not have any follow-up on this case.
-
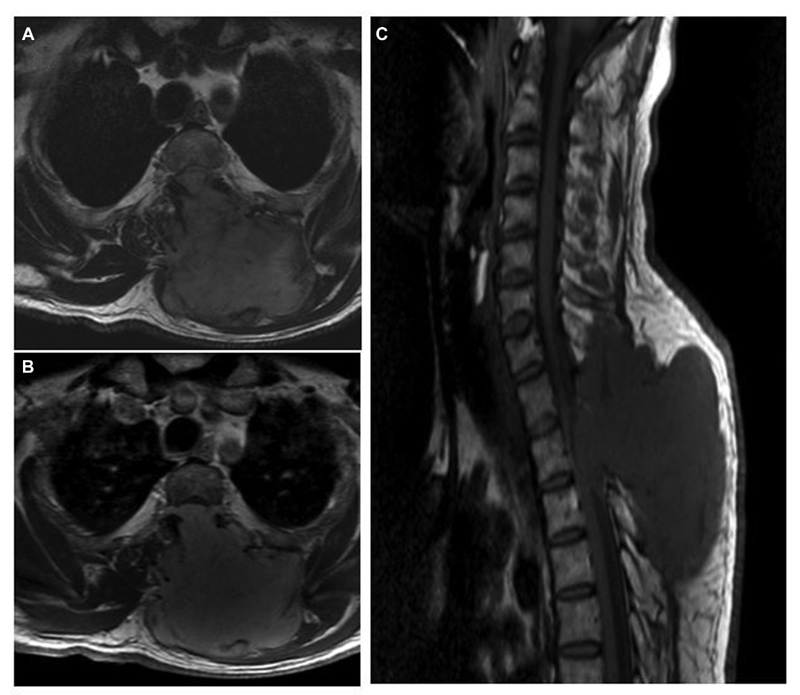
Fig. 6 Spine magnetic resonance imaging of left-side C7-D3 extradural malignant peripheral nerve sheath tumor. (A) T2-weighted axial image showing dumb-bell-shaped lesion with small intraspinal component and intramuscular part with bony destruction of the lamina and spinous process. (B) Contrast-enhanced axial image showing enhancing tumor extending into the subcutaneous region. (C) Sagittal T1-weighted image showing anteriorly compressed cord due to an intraspinal extradural component of the lesion.
Fig. 6 Spine magnetic resonance imaging of left-side C7-D3 extradural malignant peripheral nerve sheath tumor. (A) T2-weighted axial image showing dumb-bell-shaped lesion with small intraspinal component and intramuscular part with bony destruction of the lamina and spinous process. (B) Contrast-enhanced axial image showing enhancing tumor extending into the subcutaneous region. (C) Sagittal T1-weighted image showing anteriorly compressed cord due to an intraspinal extradural component of the lesion.
Case 6
A 30-year-old female presented with backache, complete weakness, numbness of both legs for 6 months, and urinary retention for 15 days. Her MRI spine showed an intradural extramedullary lesion located anterior and left of the cord extending from D6 (dorsal 6) to L2 (Lumbar 2) level. It was hypointense to isointense on T1W and T2W images with heterogenous contrast enhancement on MRI. She underwent a D5 to L2 laminotomy and near-total resection of the lesion. The histopathologic examination showed cellular spindle cell neoplasm with prominent Schwannian features but with very high mitosis (9/10 HPF) and large multifocal areas of geographic necrosis compatible with high-grade MPNST. She did not receive any form of adjuvant therapy due to financial reasons. But to our surprise, even after 7 years after surgery, she is alive with the same neurologic deficits without any radiological recurrence.
Case 7
A 16-year-old girl presented with swelling over the mid-back region for 3 months and weakness of both lower limbs for 3 days. She had a history of pulmonary tuberculosis in childhood, for which she had taken a complete course of antitubercular therapy. Her MRI dorsal spine showed a right-sided large paraspinal lesion, which extended through the neural foramina at D5 vertebral level into the vertebral canal, compressing and displacing the spinal cord to the left side. The lesion was well defined, hypointense on T2W images, and isointense on T1W images and was heterogeneously enhancing on contrast (Fig. 7). She underwent complete excision of the paraspinal and intraspinal lesion through the posterior midline approach and D3 (dorsal 3) to D6 (dorsal 6) laminoplasty. Histology of the tumor was typical of MPNST and confirmed by immunohistochemistry. There was no neurological worsening after surgery, and on follow-up, she had persistent spasticity in the lower limbs with walking difficulty. She underwent resurgery twice for significant radiological recurrence and clinical worsening at a different center after 3 and 6 months of initial surgery. Despite adjuvant radiotherapy following the third surgery, she succumbed within 6 months of third surgery and 16 months after diagnosis.
-
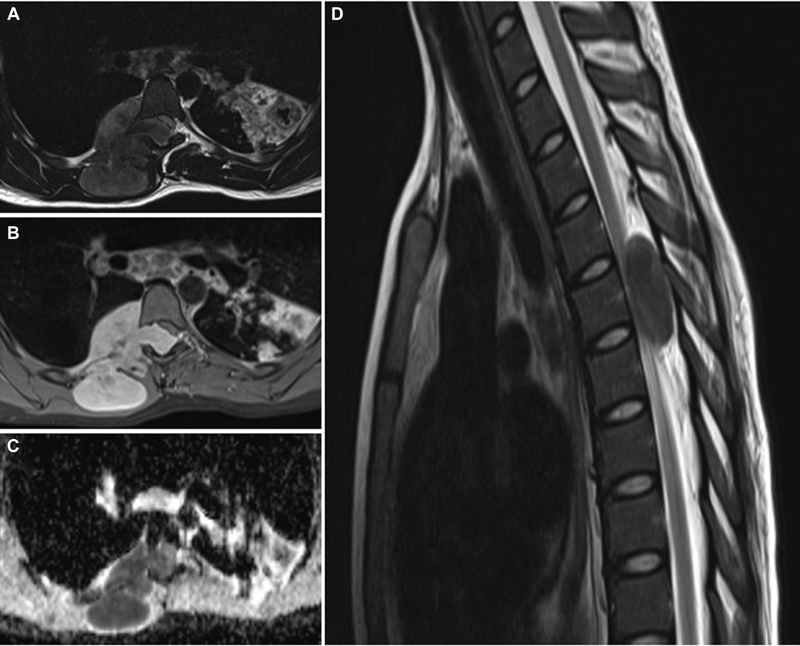
Fig. 7 Spine magnetic resonance imaging of right-side dumb-bell shaped D5 extradural malignant peripheral nerve sheath tumor. (A) T2-weighted (T2W) axial image showing large extradural dumb-bell-shaped tumor, entering intraspinal through D5 neural foramina. The lesion was predominantly hyperintense with anterolateral extension to the thoracic cavity and posterior extension in the subcutaneous region. (B) Contrast-enhanced axial image showing intensely enhancing lesion with the destruction of rib anteriorly and invasion of paraspinal muscles. However, dura and pleura appear intact. (C) Apparent diffusion coefficient (ADC) axial image showing lesion with low ADC values, suggesting facilitated diffusion. (D) T2W sagittal image of the cervicodorsal spine showing small intraspinal hyperintense lesion compressing the spinal cord entirely.
Fig. 7 Spine magnetic resonance imaging of right-side dumb-bell shaped D5 extradural malignant peripheral nerve sheath tumor. (A) T2-weighted (T2W) axial image showing large extradural dumb-bell-shaped tumor, entering intraspinal through D5 neural foramina. The lesion was predominantly hyperintense with anterolateral extension to the thoracic cavity and posterior extension in the subcutaneous region. (B) Contrast-enhanced axial image showing intensely enhancing lesion with the destruction of rib anteriorly and invasion of paraspinal muscles. However, dura and pleura appear intact. (C) Apparent diffusion coefficient (ADC) axial image showing lesion with low ADC values, suggesting facilitated diffusion. (D) T2W sagittal image of the cervicodorsal spine showing small intraspinal hyperintense lesion compressing the spinal cord entirely.
Discussion
The histopathological diagnosis of this enigmatic entity was first reported almost 70 years ago, but no uniform diagnostic criteria are available in terms of clinical or radiological features. In 1952, Cuneo and Rand reported the first case of MPNST,14 while intracranial MPNST was reported after 30 years in 1983 by Kudo et al when they reported the first case of MPNST arising from the VIIIth nerve.15 The term MPNST was coined by the World Health Organization (WHO) to replace various names that were in use previously, like “malignant schwannoma,” “neurofibrosarcoma,” “neurogenic sarcoma,” and “malignant neurilemmoma.” They arise from the healthy peripheral nerve or the pre-existing nerve sheath tumors. Overall, the incidence of MPNST is as low as 0.01%.2 Intracranial spinal MPNST is very rare, and hence many of the treatment strategies are based on our knowledge about their peripheral counterparts.
These tumors arise de novo from healthy peripheral nerve (50%) or de-differentiation from pre-existing schwannoma or neurofibromas. There is a strong association between MPNST and NF1 (13%) or NF2 (6%). The common intracranial nerves to get involved are the VIIIth nerve (60%), the Vth nerve (27%), or the VIIth nerve (10%).12 In our series, three out of four intracranial cases (75%) had cranial nerve MPNST, two involving the Vth nerve and one involving the VIIIth nerve. The secondary MPNST occurs by either dedifferentiation of pre-existing lesions spontaneously or in some studies following ionizing radiation. It may cause chromosomal aberrations and induce cellular atypia in pre-existing schwannomas leading to malignant degeneration in as many as 41.7% of cases.8 16 However, one of the recent articles on long-term follow-up of various pathologies following GKRS did not support the idea of single-fraction radiation leading to malignant degeneration.17 There are some reports of intraparenchymal MPNST without any association with the cranial nerves. The histogenesis remains controversial. Neuropathologists believe that they arise from perivascular nerves descending into the parenchyma or spinal cord.18 19 MPNST is known for its very aggressive behavior with rapid regrowth and metastasis.20
Epidemiology
Due to a very low incidence of the lesion and lack of available literature, most of them initially get misdiagnosed as benign lesions and, on rare occasions, get labeled as other malignant tumors like glioblastoma, as in one of the cases in our series. In the present case series of seven MPNST patients, we reported a diverse variety of cranial and spinal MPNST along the neuroaxis (Table 1). Three cases were associated with cranial nerves in the four cranial cases, and another was an intraparenchymal lesion. In the three spinal cases of MPNST, two were extradural, and one was intradural extramedullary. In none of the cases, we suspected MPNST preoperatively based on the clinical scenario or radiological findings. The diagnosis was confirmed only after histopathologic examination.
|
Age/sex |
Location |
Precursor |
RT |
NF |
Radiological features |
Surgery |
Chemotherapy |
RT |
Follow-up |
Present status |
|---|---|---|---|---|---|---|---|---|---|---|
|
52/ F |
VIIIth |
Schwannoma |
N |
NF2 |
Ill-defined lesion, perilesional edema. |
STR |
N |
Y |
11 mo |
Alive |
|
34/ F |
Temporal |
De novo |
N |
N |
Ill-defined lesion, heterogenous enhancement, high Ch:Cr ratio. |
GTR |
Y |
Y |
48 mo |
Death |
|
39/ F |
Vth |
Schwannoma |
Y (GKRS) |
N |
Ill-defined lesion, heterogenous enhancement |
GTR |
N |
Y |
8 mo |
Alive |
|
18/ M |
Vth |
De novo |
N |
N |
Large lesion, heterogeneous enhancement, diffusion restriction |
STR |
N |
Y |
14 mo |
Alive |
|
51/ M |
Dorsal |
De novo |
N |
N |
Long-segment disease, bony destruction |
GTR |
NA |
NA |
Lost |
NA |
|
30/ F |
Dorsolumbar |
De novo |
N |
N |
Long-segment disease, heterogeneous enhancement |
NTR |
N |
N |
84 mo |
Alive |
|
16/ F |
Dorsal |
De novo |
N |
N |
Large lesion, heterogeneous enhancement |
GTR |
N |
Y |
16 mo |
Death |
Abbreviations: F, female; GTR, gross total resection; M, male; mo, month; MPNST, malignant peripheral nerve sheath tumor; N, no; NA, not applicable; NF, neurofibromatosis; NTR, near-total resection; RT, radiation therapy; STR, subtotal resection; Vth, trigeminal nerve; Y, yes.
To the best of our knowledge, we could find 62 cases of craniospinal MPNST reported in the literature to date. Out of these, only 17 cases have been intraparenchymal lesions, while others arise from one of the nerves, either cranial or spinal.21 22 Although the epidemiological data are very minimal, few literature reviews suggest that the average age is 39 years, with a male-to-female ratio of 1.5:1.23 In our short series, the average age of the cohort was 34.3 years, with a male to female ratio of 0.2:1. Five out of seven (71.43%) tumors presented de novo, and we could achieve GTR in four out of seven (57.14%) patients. In NF2 with bilateral schwannoma and Vth nerve schwannoma extending extracranially, we could achieve STR.
Radiological Features
The MRI features cannot make a clear distinction between malignant and benign nerve sheath tumors. Size higher than 5 cm, infiltrative margins, and heterogenous appearance is likely to clinch the diagnosis of MPNST over benign variety.5 Size criteria can be challenging to apply in some CNS cases. Other features like ill-defined margins, invasion of adjacent structures with peritumoral edema,24 intratumoral cystic/necrotic/hemorrhagic changes,25 nodular morphology, or inhomogeneity,26 and frequent intratumoral calcifications21 can be helpful to differentiate malignant versus benign lesions. Also, invasion of adjacent structures, destruction of surrounding bone by pressure, or frank destruction is seen in 20% of cases, and it favors the diagnosis of MPNST.21 Demeheri et al reported that the minimum apparent diffusion coefficient is lower in malignant tumors than benign MPNSTs.24 The dynamic contrast-enhanced imaging and MR spectroscopy are additional MR sequences that can help differentiate these tumors from their benign counterparts. Spinal MRI should ideally be performed before surgical intervention in all intracranial cases as drop metastases are frequently seen in the spine. Our case series found that most of our malignant tumors were hypointense or isointense on T2-weighted images. While in literature, benign counterparts are commonly hyperintense in the same sequence. The other criteria reported in the literature, like ill-defined lesions, invasiveness, perilesional edema, and large size, match our cohort's imaging findings.
Pathological and Immunohistochemical Features, Grading
MPNST is a malignant spindle cell, nerve sheath tumor, originating from various cell types like a peripheral cell, schwann cell, and rare fibroblasts. They arise either from a healthy nerve or from a pre-existing neoplasm like plexiform neurofibroma or schwannoma.2 7 23 27 The transition of MPNST from neurofibroma is expected at the peripheral location, while at intracranial locations, the change from schwannoma is more common.23 Histologically, MPNST has varied morphology ranging from low grade to high grade (Fig. 6). They exhibit fasciculated patterns, herringbones patterns, and hemangiopericytoma-like areas. Rarely intracranial MPNST shows a divergent differentiation like a skeletal muscle.28 Specific biomarkers help highlight transformation into MPNST, such as loss of expression of SOX10, S-100, CD34, H3k27Me3, and CDKN2A. The Ki67 proliferative index >10% and strong P53 positivity suggest MPNST.29 Most of our cases exhibited patchy S100 staining with a high Ki67 labeling index. H3 k27me3 performed in one patient showed a loss of expression. P53 staining is high in two of the three cases tested. Currently, peripheral and intracranial MPNSTs are graded into low and high-grade based on mitosis and necrosis. High-grade tumors have mitotic count >4/HPF and necrosis.30 Histopathology remains a vital cog in the diagnosis because soft tissue sarcomas contain heterogeneous areas, leading to discordance between different pathologists. There is no such staging system at present for intracranial MPNSTs.13
Treatment
As with the diagnosis, even the treatment of craniospinal MPNSTs still lacks good evidence to make a definitive policy. The established treatment protocols are based on knowledge about peripheral MPNSTs. The mainstay of the treatment is radical surgical resection. The studies by Anghileri et al and Zou et al confirmed that complete resection improves overall survival and is inversely related to the local recurrence.9 10 16 Due to the difficult locations of intracranial MPNSTs, sometimes complete resection is not feasible, and partial resection followed by adjuvant radiotherapy can be considered a helpful alternative.
It is difficult to fathom the role of adjuvant radiotherapy as part of a treatment considering that ionizing radiation has been implicated in the pathogenesis of some cases. Gousias et al evaluated the factors influencing survival and confirmed the positive impact of radiation therapy on overall survival. In the univariate analysis, they found 50 to 60 Gy had a positive effect on survival, but the same did not hold on multivariate analysis.16 In an institutional study by Anghileri et al on prognostic factors, more prolonged overall survival was noted on multivariate analysis with adjuvant radiotherapy. Still, the correlation between local recurrence and distant metastasis was not seen.9 The radiation dose is not uniform across various studies, but 60 Gy is routinely prescribed.16 There is further controversy on the role of chemotherapy in MPNST, as these tumors do not respond to chemotherapy very well.3 12 Although peripheral MPNSTs failed to show any survival benefit with adjuvant chemotherapy,11 31 combined radiochemotherapy after surgery improved survival in a study by Minovi et al on cases of head and neck MPNSTs.32 Doxorubicin had been the gold standard in chemotherapy treatment for soft tissue sarcomas.33 Recent trials EORTC 62012 comparing doxorubicin alone versus doxorubicin plus ifosfamide for locally advanced unresectable or metastatic soft-tissue sarcomas showed patients with high-grade tumors, and worse performance status benefited the most. Still, no improvement in overall survival, so it does not support the use of combination chemotherapy for palliation unless the specific goal is tumor shrinkage.34 In 2017, subgroup analysis of this trial showed doxorubicin remains the standard of care first-line chemotherapy for advanced soft tissue sarcomas, and combination therapy appropriate for selected patients, age < 60 years and poorly differentiated tumors.33
Compared with benign PNSTs like schwannomas and neurofibromas, MPNSTs have a poor prognosis with mean overall survival of 19.3 months3 and a median overall survival of 9 months (range: 1–66 months). The 5-year mortality rate of CNS or peripheral MPNSTs is 56 to 67%.9 10 Our series has an overall survival rate of 57.14%. The most important and probably the only factor affecting the overall survival is the extent of resection.16 The factor that adversely affects survival is previous radiation exposure, which leads to malignant degeneration in the benign lesion and restricts the therapeutic options for clinicians.23
As these tumors have a lower survival rate with a high chance of recurrence and metastasis, close clinical and radiological follow-up is mandatory. In the first-year post-radiotherapy, radiological follow-up was done every 6 months. It includes cranial and spinal MRI also, as, in some studies, spinal metastases have been common,12 along with clinical evaluation. After the first year of monitoring, visits can be at every 6 months and can be continued lifelong. Although the treatment strategy for recurrence is still debatable, routine oncological principles of treatment for recurrence, as for any other CNS malignancy, can be applied at present until further evidence is available on the topic.
Conclusions
MPNST is a rare intracranial malignancy. They are commonly associated with NF1 or NF2 with malignant degeneration of precursor lesions, but most of them arise as de novo lesions. Prior history of radiation exposure is one of the reported etiological factors for malignant transformation.
Pathological confirmation can be a challenge, as with radiology. But it remains the cornerstone in the diagnostic armamentarium of clinicians. Shorter history and rapid growth with heterogeneous invasive appearance are certain clues to be kept in mind before reaching the diagnosis.
The aim of treatment should be maximal resection followed by adjuvant radiotherapy. Close postoperative follow-up, both clinically and radiologically with craniospinal imaging, is crucial in the early detection of recurrence.
Conflict of Interest
None declared.
Funding None.
References
- Malignant soft-tissue tumors in a large referral population: distribution of diagnoses by age, sex, and location. AJR Am J Roentgenol. 1995;164(1):129-134.
- [Google Scholar]
- Malignant peripheral nerve sheath tumors. A clinicopathologic study of 28 cases. Cancer. 1993;71(4):1247-1253.
- [Google Scholar]
- Malignant peripheral nerve sheath tumors. A clinicopathologic study of 120 cases. Cancer. 1986;57(10):2006-2021.
- [Google Scholar]
- Tumors and tumor-like lesions of peripheral nerves. Semin Musculoskelet Radiol. 2010;14(5):547-558.
- [Google Scholar]
- The cytopathology of malignant peripheral nerve sheath tumor: a report of 55 fine-needle aspiration cases. Cancer Cytopathol. 2012;120(5):334-341.
- [Google Scholar]
- Schwannoma (neurilemoma) with malignant transformation. A rare, distinctive peripheral nerve tumor. Am J Surg Pathol. 1994;18(9):882-895.
- [Google Scholar]
- Radiation-induced malignant and atypical peripheral nerve sheath tumors. Ann Neurol. 1980;7(4):311-318.
- [Google Scholar]
- Malignant peripheral nerve sheath tumors: prognostic factors and survival in a series of patients treated at a single institution. Cancer. 2006;107(5):1065-1074.
- [Google Scholar]
- Clinical, pathological, and molecular variables predictive of malignant peripheral nerve sheath tumor outcome. Ann Surg. 2009;249(6):1014-1022.
- [Google Scholar]
- Diagnosis and management of intracranial malignant peripheral nerve sheath tumors. Neurosurgery. 2008;62(4):825-832. , discussion 832
- [Google Scholar]
- Malignant peripheral nerve sheath tumor of intracranial nerve: a case series review. Auris Nasus Larynx. 2010;37(5):539-545.
- [Google Scholar]
- Predictive and prognostic factors associated with soft tissue sarcoma response to chemotherapy: a subgroup analysis of the European Organisation for Research and Treatment of Cancer 62012 study. Acta Oncol. 2017;56(7):1013-1020.
- [Google Scholar]
- Tumors of the gasserian ganglion; tumor of the left gasserian ganglion associated with enlargement of the mandibular nerve; a review of the literature and case report. J Neurosurg. 1952;9(5):423-431.
- [Google Scholar]
- Malignant nerve sheath tumor of acoustic nerve. Arch Pathol Lab Med. 1983;107(6):293-297.
- [Google Scholar]
- Factors of influence upon overall survival in the treatment of intracranial MPNSTs. Review of the literature and report of a case. Radiat Oncol. 2010;5:114.
- [Google Scholar]
- The risk of radiation-induced tumors or malignant transformation after single-fraction intracranial radiosurgery: results based on a 25-year experience. Int J Radiat Oncol Biol Phys. 2017;97(5):919-923.
- [Google Scholar]
- Malignant intracerebral nerve sheath tumor: a case report and review of the literature. Cancer. 1998;82(3):545-552.
- [Google Scholar]
- Malignant intracerebral nerve sheath tumor with intratumoral calcification. Case report. J Neurosurg. 2000;92(2):338-341.
- [Google Scholar]
- Malignant intraventricular schwannoma. Case report. J Neurosurg. 1995;82(1):121-124.
- [Google Scholar]
- CT, MRI, and 18F-FDG PET/CT findings of malignant peripheral nerve sheath tumor of the head and neck. Acta Radiol. 2017;58(10):1222-1230.
- [Google Scholar]
- Malignant peripheral nerve sheath tumor of the oral cavity. J Oral Maxillofac Surg. 2012;70(10):e581-e585.
- [Google Scholar]
- Updates on the diagnosis and treatment of intracranial nerve malignant peripheral nerve sheath tumors. OncoTargets Ther. 2013;6:459-470.
- [Google Scholar]
- Conventional and functional MR imaging of peripheral nerve sheath tumors: initial experience. AJNR Am J Neuroradiol. 2014;35(8):1615-1620.
- [Google Scholar]
- MRI features in the differentiation of malignant peripheral nerve sheath tumors and neurofibromas. AJR Am J Roentgenol. 2010;194(6):1568-1574.
- [Google Scholar]
- The value of magnetic resonance imaging in the differentiation between malignant peripheral nerve-sheath tumors and non-neurogenic malignant soft-tissue tumors. Skeletal Radiol. 2006;35(10):745-753.
- [Google Scholar]
- Malignant peripheral nerve sheath tumors of cranial nerves and intracranial contents: a clinicopathologic study of 17 cases. Am J Surg Pathol. 2009;33(3):325-338.
- [Google Scholar]
- A rare case of intracranial malignant triton tumor arising in the middle cranial fossa: a case report and review of the literature. Folia Neuropathol. 2018;56(3):229-234.
- [Google Scholar]
- Neurofibromatosis type 1-associated MPNST state of the science: outlining a research agenda for the future. J Natl Cancer Inst 2017;109(08):djx124
- [Google Scholar]
- Pathology of peripheral nerve sheath tumors: diagnostic overview and update on selected diagnostic problems. Acta Neuropathol. 2012;123(3):295-319.
- [Google Scholar]
- Surgical management of malignant peripheral nerve sheath tumors. Neurosurg Focus. 2007;22(6):E13.
- [Google Scholar]
- Malignant peripheral nerve sheath tumors of the head and neck: management of 10 cases and literature review. Head Neck. 2007;29(5):439-445.
- [Google Scholar]
- First-line chemotherapy for malignant peripheral nerve sheath tumor (MPNST) versus other histological soft tissue sarcoma subtypes and as a prognostic factor for MPNST: an EORTC soft tissue and bone sarcoma group study. Ann Oncol. 2011;22(1):207-214.
- [Google Scholar]
- Doxorubicin alone versus intensified doxorubicin plus ifosfamide for first-line treatment of advanced or metastatic soft-tissue sarcoma: a randomised controlled phase 3 trial. Lancet Oncol. 2014;15(4):415-423.
- [Google Scholar]




