
Translate this page into:
Comparison of clinico-radiological profile, optical coherence tomography parameters, and outcome in MOGAD and Neuromyelitis optica spectrum disorder subtypes: A prospective observational study
*Corresponding author: Niraj Kumar, Department of Neurology, AIIMS, Rishikesh, Uttarakhand, India. drnirajkumarsingh@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Dhar N, Kumar M, Tiwari A, Samanta R, Bhadoria AS, Vivekanandhan S, et al. Comparison of clinico-radiological profile, optical coherence tomography parameters, and outcome in MOGAD and Neuromyelitis optica spectrum disorder subtypes: A prospective observational study. J Neurosci Rural Pract 2023;14:239-51.
Abstract
Objectives:
The objectives of the study were to compare the clinico-radiological profile, optical coherence tomography (OCT) parameters and outcome in Myelin Oligodendrocyte Glycoprotein-IgG-associated disorders (MOGAD) and Neuromyelitis Optica Spectrum disorder subtypes.
Materials and Methods:
This prospective study involved collection of data regarding neurological assessment, neuroimaging, cerebrospinal fluid analysis, OCT parameters, treatment and outcome. Disease severity and disability were assessed using Expanded Disability Status Scale and modified Rankin scale. Patients were categorized into aquaporin-4 (AQP4+), MOGAD, and double negative (DN; both AQP4 and MOG negative).
Results:
Among 31 patients included, 42% were AQP4+, 32.2% were MOGAD, and 25.7% were DN. The median age at onset was comparable (AQP4+ vs. MOGAD vs. DN = 28 years vs. 24.4 years vs. 31.5years; P = 0.31). Females predominated in AQP4+ compared to MOGAD group (76.9% vs. 30%; P = 0.02). Majority of patients (73.5%) had a relapsing course with a median of two (range = 1–9) relapses. Ninety-nine demyelinating events occurred: Transverse myelitis (TM) in 60/99 (60.6%), optic neuritis (ON) in 43/99 (43.4%), area postrema (AP) syndrome in 20/99 (20.1%), and optico-spinal syndrome in 10/99 (10.1%). ON was common in MOGAD than AQP4+ patients (58.6% vs. 32.1%; P = 0.03). Spinal cord and brain lesions on magnetic resonance imaging (MRI) were seen in 90.3% and 54.8% patients, respectively. A significantly higher proportion of AQP4+ patients showed longitudinally extensive transverse myelitis as compared to MOGAD group (69.2 % vs. 20 %; P = 0.04), specifically involving dorsal cord (92.3% vs. 50%; P = 0.02). MRI brain lesions, especially involving AP, was frequent in DN than MOGAD (47.1% vs. 6.9%; P = 0.003) and AQP4+ (47.1% vs. 18.9%; P = 0.03) patients. AQP4+ group showed significant nasal RNFL thinning on OCT (P = 0.04). Although 6-month good functional outcome was better in MOGAD than DN and AQP4+ (80% vs. 71.4% vs. 41.7%) groups, they were comparable (P = 0.13).
Conclusion:
Nearly three-fourth of our patients showed a relapsing course, with TM being the most common clinical presentation. AQP4+ group showed female preponderance, frequent dorsal cord longitudinally extensive transverse myelitis, less frequent ON, and greater nasal RNFL thinning compared to MOGAD group. MRI brain lesions were more common in DN patients. All three groups exhibited good response to pulse corticosteroids and showed a comparable functional outcome at 6-month follow-up.
Keywords
Neuromyelitis optica spectrum disorder
Optical coherence tomography
Myelin oligodendrocyte glycoprotein
Longitudinally extensive transverse myelitis
Optic neuritis

INTRODUCTION
Myelin oligodendrocyte glycoprotein-IgG-associated disorders (MOGAD), aquaporin 4-IgG-seropositive (AQP4+) neuromyelitis optica spectrum disorder (NMOSD), and double negative (DN) patients, having both AQP4 and MOG-antibody negative, form a group of rare inflammatory disorders of the central nervous system (CNS). They are characterized by simultaneous or rapidly sequential immune-mediated demyelination, with a predilection for optic nerve and spinal cord. The prevalence of this group of disorders ranges from 3.9 to 4.4/100,000 in the Western World, with a higher prevalence 10/100,000 seen in AfroCaribbeans/African Americans.[1] A prevalence of 2.6/100,000 has been reported in an Indian study using a registry method.[2] While this group of CNS disorder predominantly comprises of AQP4+ and MOGAD patients, nearly 16% patients remain DN.[3] Besides neuroimaging and serological testing, spectral domain-optical coherence tomography (SD-OCT), which measures retinal nerve fiber layer thickness (RNFLT), may help distinguish various patterns of demyelinating disorders in patients with optic neuritis (ON).[4]
It is paramount to differentiate MOGAD and NMOSD subtypes from multiple sclerosis (MS), as therapeutic approaches differ and lack of optimal therapy can result in significant disability including permanent blindness and paralysis. To date, only a handful of studies[5-16] have evaluated MOGAD and NMOSD subtypes in India, with half of them having a retrospective design.[5-7,9,10,15] Only a couple of Indian studies have discussed the characteristics of DN subtype.[6,13] Moreover, none of these Indian studies have compared RNFLT patterns in MOGAD and NMOSD subtypes. Herein, we compare the RNFLT in addition to the clinico-radiological and laboratory parameters along with outcome in MOGAD and NMOSD subtypes.
MATERIALS AND METHODS
This prospective observational study included 31 patients managed at a University Hospital from December 2019 to January 2021. The study protocol was approved by Institutional Ethics Committee (AIIMS/IEC/19/1257).
Inclusion and exclusion criteria
All consenting patients satisfying the International Panel for NMO Diagnosis -2015 criteria for NMOSD[17] and/or fulfilling the diagnostic criteria for MOGAD with anti-MOG antibody positivity (by cell-based assay)[18] were included. Patients with MS or secondary demyelination due to vascular, infectious, and/or immunological causes were excluded from the study. Also excluded were those with prior intraocular surgery or laser treatment, glaucoma, retinal diseases, and diabetes.
Data collection
Included patients underwent detailed neurological and ophthalmological evaluation, cerebrospinal fluid (CSF) analysis, contrast-enhanced magnetic resonance imaging of brain, orbit, and spine (using 3-T magnetic resonance imaging [MRI] General Electric machine), visual evoked potentials, and brainstem auditory evoked response. AQP4 and MOG antibodies were tested by fixed cell-based indirect immunofluorescence assay (using EUROIMMUNE Kit). Both eyes of each patient were evaluated for best corrected visual acuity (distant and near vision), fundoscopy and SDOCT to measure RNFLT, average macular thickness and volume. Assessment of RNFLT and macular imaging was done using CIRRUS HD-OCT 500 (Carl Zeiss Meditec Inc, Dublin, CA, USA) after dilation with 1% tropicamide. The RNFLT was studied in the superior (S), temporal (T), nasal (N) and inferior (I) quadrants. The average global thickness (G) was extrapolated from the formula (G = [S + N + T + I]/4). All patients were given a visual functional severity score (VFSS) based on their visual acuity (rated from 0 to 6) at admission, 3 and 6-month follow-up assessment [Supplementary Table 1]. All visual acuity data were converted to logarithm of the minimum angle of resolution for statistical analysis. Patients who could only count fingers, perceive hand movements close to face, had only light perception, or had no light perception were assigned the values of 2.6, 2.7, 2.8, and 2.9, respectively.[19] Disease severity and disability were documented in terms of Modified Rankin Scale (mRS)[20] and Expanded Disability Status Scale (EDSS).[21]
Treatment including both acute and maintenance therapy and outcome details were recorded. The follow-up assessments at 3 and 6 months and/or whenever indicated clinically, included recording therapeutic response in terms of number of relapses, change in EDSS score and requirement of steroid sparing (long-term treatment) agents. Moreover, at each follow-up visit, the dose, duration, and side effects of ongoing therapy were recorded. Management was individualized according to the severity of illness, lesion burden on neuroimaging, cost-effectiveness, patient’s past drug history, and co-morbidities. Treatment protocol for MOGAD and NMOSD subtypes is shown in [Supplementary Figure 1].[22]
Outcome measures
Primary outcome
The primary outcome of the study was to compare clinicoradiological profile and OCT parameters in MOGAD and NMOSD subtypes.
Secondary outcome
Treatment response clinically in terms of number of relapses, changes in EDSS and mRS on sequential assessment and requirement of steroid sparing agents. Good motor outcome was defined as mRS score of 0–2.
Statistical analysis
The data was analyzed using Statistical Package for the Social Sciences software (IBM SPSS Statistics for Windows, Version 23.0. Armonk, NY: IBM Corp.). Categorical variables were described as frequency and proportion. Continuous variables were expressed as mean (± standard deviation) or median (range) as applicable. Shapiro–Wilk test was used to test normality of data. Proportions were compared using Chi-square test or Fisher’s exact test. The continuous variables were compared using Student’s t-test or Mann–Whitney U test depending on the parametric or non-parametric distribution, respectively. Multiple group comparisons were done using Kruskal–Wallis/One-way ANOVA. A variable with a two-tailed P < 0•05 was considered statistically significant.
RESULTS
Of the 79 patients with acquired CNS demyelination, 31 had either MOGAD or NMOSD subtypes: 10 had MOGAD, 13 had AQP4+, and eight were DN patients. None of them had double seropositivity [Supplementary Figure 2].
Demographic and clinical features
Baseline demographic and clinical features are shown in [Table 1]. Age at disease onset and age at presentation were comparable among the three groups (P = 0.63). There was female preponderance (52.9%), with a significantly higher proportion of females in AQP4+ compared to MOGAD subtype (76.9% vs. 30%; P = 0.02). Majority of patients (73.5%) had a relapsing course with a median of two (range = 1–9) relapses. None of our patient had a progressive clinical course. Including the 68 relapses, there was a total of 99 demyelinating events in our 31 patients.
| Parameter | NMOSD whole cohort (n=31) | AQP4+(n=13) A | MOGAD (n=10) B | DN (n=8) C | P-value A versus B | P-value A versus B versus C |
|---|---|---|---|---|---|---|
| Demographic | ||||||
| Age at presentation (years): median (range) | 30 (18–61) | 39 (18–51) | 28.5 (20–61) | 32 (20–55) | 0.34 | 0.63 |
| Age at onset (years): median (range) | 27.9 (14–61) | 28 (16–50) | 24.4 (14–61) | 31.5 (18–55) | 0.26 | 0.31 |
| Female-n(%) | 17 (54.8%) | 10 (76.9%) | 3 (30%) | 4 (50%) | 0.02 | 0.08 |
| Median delay in diagnosis (months): (range) (months) | 8 (1–180) | 24 (1–180) | 13 (1–168) | 6.5 (1–36) | 0.56 | 0.22 |
| Patients with relapsing course: n(%) | 25 (73.5%) | 11 (84.6%) | 8 (80%) | 6 (54.5%) | 0.77 | 0.22 |
| Median no. of relapses (range) | 2 (1–9) | 3 (1–9) | 2 (1–8) | 2 (1–3) | 0.77 | 0.35 |
| Infectious prodrome (n)% | 3 (8.8%) | 0 | 0 | 3 (27.3%) | NA | NA |
| Co-existing autoimmunity# | 2 (5.9%) | 2 (15.4%) | 0 | 0 | NA | NA |
| Clinical features | ||||||
| First attack (n;%) | ||||||
| Isolated ON | 7 (22.6%) | 2 (15.4%) | 2 (20%) | 3 (37.5%) | >0.99 | 0.49 |
| Isolated TM | 12 (38.7%) | 4 (30.8%) | 5(50%) | 3 (37.5%) | 0.58 | 0.42 |
| Isolated AP | 2 (6.5%) | 2 (15.4%) | 0 | 0 | – | – |
| ON+TM | 3 (9.7%) | 1 (7.7%) | 2(20%) | 0 (0.0%) | 0.77 | 0.91 |
| Others* | 7 (22.6%) | 4 (30.8%) | 1(10%) | 2 (25%) | 0.34 | 0.49 |
| Associated features | ||||||
| PPTS | 4 (11.8%) | 4 (30.8%) | 0 | 0 | NA | NA |
| Headache | 6 (17.6%) | 2 (15.4%) | 2 (20%) | 2 (25%) | 0.77 | 0.96 |
| Seizure | 1 (2.9%) | 0 | 0 | 1 (12.5%) | NA | NA |
Clinical characteristics of demyelinating episodes at onset
Isolated transverse myelitis (TM) was the most common presentation at onset in the entire cohort (38.7%) as well as in AQP4+ (30.8%), MOGAD (50%), and DN (37.5%) groups. ON, isolated, or in combination with other symptoms, was seen in 5 (38.5%), 4 (40.0%), and 5 (62.5%) patients (P = 0.52), of which 3 (60%), 2 (50.0%), and 3 (60%) patients (P = 0.94) had bilateral ON in AQP4+, MOGAD, and DN groups, respectively. Isolated area postrema (AP) syndrome was seen in 2 (15.4%) patients with AQP4+ and none with MOGAD and DN.
Clinical characteristics of relapses
In the 68 relapses seen in 31 patients, isolated TM was the most common presentation in the entire cohort (42.6%) as well as in AQP4+ (47.5%) and DN (44.4%) groups. Isolated ON (42.1%) was the most frequent manifestation during the relapses in MOGAD [Supplementary Table 2].
Clinical characteristics of all demyelinating episodes
Of the 99 demyelinating events in our cohort, 53 occurred in AQP4+, 29 in MOGAD and 17 in DN group. The common clinical presentations included TM (60/99 = 60.6%), ON (43/99 = 43.4%), AP syndrome (20/99 = 20.1%), and optico-spinal syndrome (10/99 = 10.1%) [Supplementary Figure 3]. The overall incidence of each clinical presentation was similar among the three groups, with subtle differences observed in the incidences of ON and AP syndrome. ON comprised a significantly higher proportion of demyelinating events in MOGAD as compared to AQP4+ patients (58.6% vs. 32.1%; P = 0.03); however, it was comparable to DN group (58.6% vs. 52.9%; P = 0.76). Frequency of AP syndrome was significantly higher in DN patients as compared to MOGAD (47.1% vs. 6.9%; P = 0.003) and AQP4+ (47.1% vs. 18.9%; P = 0.03) patients. AQP4+ and DN patients were more likely to develop involvement of other areas including diencephalon and cerebral cortex, as seen in 3.7% and 17.6%, respectively, compared to none in MOGAD group.
Neuroimaging and laboratory features
Spinal cord and brain lesions on MRI were seen in 90.3% and 54.8% of patients, respectively. Cervical and dorsal regions were the commonly involved sites in the spinal cord. A significantly higher proportion of AQP4+ patients developed longitudinally extensive transverse myelitis (LETM) as compared to MOGAD group (69.2% vs. 20%; P = 0.04), specifically for dorsal cord involvement (92.3% vs. 50%; P = 0.02). Optic nerve lesions were seen in 18 (58%) patients. While posterior optic nerve, including chiasma, was more commonly involved in AQP4+ patients compared to MOGAD (46.1% vs. 10%; P = 0.01), anterior optic nerve (particularly intracanalicular portions) involvement was more common in the latter group compared to former (70% vs. 30.8%; P = 0.06). [Figures 1 and 2] show some of the MRI images of our patients. [Table 2] summarizes the distribution of MRI lesions along with serological and CSF study results.

- Spinal cord and optic nerve lesions (arrows) on magnetic resonance imaging (MRI) in Myelin Oligodendrocyte Glycoprotein-IgG-associated disorders and neuromyelitis optica spectrum disorder subtypes, MRI spine T2W sagittal (a) image showing longitudinally extensive hyperintense signal in dorsal cord in a patient with Aquaporin 4 antibody positive patients (AQP4) antibody positive disease, with the T2W axial image shown in panel (b). MRI spine T2W sagittal (c) image showing patchy hyperintensity in the cervicodorsal cord in a patient with MOG antibody positive disease, with the T2W axial image shown in panel (d). MRI orbit T2W axial (e) image showing left anterior segment hyperintense signal in the optic nerve in a patient with MOG antibody positive disease. MRI orbit T1W with Gadolinium axial (f) image showing bilateral posterior segment hyperintense signal in the optic nerve in a patient with AQP4 antibody positive disease.

- Brainstem lesions (arrows) on magnetic resonance imaging (MRI) of a patient Aquaporin 4 antibody positive patients (AQP4) antibody positive disease, MRI brain T2W axial (a) image showing hyperintense signal in dorsal part of medulla in a patient with positive AQP4 antibody, with the T2W sagittal image seen in panel (b).
| Parameter | AQP4+(n=13) A (%) | MOGAD (n=10) B (%) | DN (n=8) C (%) | P-value A versus B versus C | P-value A versus B |
|---|---|---|---|---|---|
| MRI Optic nerve | |||||
| Unilateral | 1 (7.7) | 3 (30) | 3 (37.5) | 0.77 | 0.77 |
| Bilateral | 5 (38.5) | 4 (40) | 2 (25) | ||
| Intraorbital | 3 (23.1) | 5 (50) | 337.5) | 0.41 | 0.18 |
| Intracanalicular | 4 (30.8) | 7 (70) | 2 (25) | 0.09 | 0.06 |
| Intracranial | 7 (53.8) | 4 (40) | 2 (25) | 0.42 | 0.51 |
| Chiasmal | 6 (46.1) | 1 (10) | 2 (25) | 0.05* | 0.01* |
| Tract | 2 (15.4) | 0 | 1 (12.5) | NA | NA |
| Intraorbital | 3 (23.1) | 5 (50) | 3 (37.5) | 0.41 | 0.18 |
| MRI Brain | |||||
| Diencephalon | 0 | 0 | 2 (25) | NA | NA |
| Area postrema | 2 (15.4) | 3 (30) | 4 (50) | 0.24 | 0.36 |
| Periventricular | 2 (15.4) | 3 (30) | 4 (50) | 0.24 | 0.36 |
| Corona radiata (WM) | 2 (15.4) | 2 (20) | 3 (37.5) | 0.49 | 0.59 |
| Periependymal | 1 (7.7) | 2 (20) | 0 | NA | 0.39 |
| Corpus callosum | 0 | 1 (10) | 3 (37.5) | NA | NA |
| Type of Spinal involvement | |||||
| Multiple short segments | 4 (30.8) | 6 (60) | 2 (25) | 0.24 | 0.21 |
| LETM | 9 (69.2) | 2 (20) | 4 (50) | 0.06 | 0.04* |
| Site of spinal involvement | |||||
| Cervical | 11 (84.6) | 7 (70) | 4 (50) | 0.24 | 0.37 |
| Dorsal | 12 (92.3) | 5 (50) | 6 (75) | 0.08 | 0.02* |
| Lumbar | 4 (30.8) | 3 (30) | 1 (12.5) | 0.61 | 0.87 |
| CSF analysis | |||||
| Pleocytosis (>5 cells/cm3) | 3 (23.1) | 5 (50) | 3 (37.5) | 0.18 | 0.42 |
| Elevated Proteins (>50 mg/dL) | 10 (76.9) | 10 (100) | 5 (62.5) | 0.16 | 0.13 |
| Presence of OCB | 2 (15.4) | 1 (10) | 1 (12.5) | 0.61 | 0.93 |
| Serum analysis | |||||
| High ACE levels (>40 U/L) | 0 (0) | 1 (10) | 3 (37.5) | NA | 0.05 |
| ANA (>1:100) | 2 (15.4) | 0 (0) | 1 (12.5) | NA | 0.49 |
ACE: Angiotensin converting enzyme, ANA: Anti-nuclear antibody, AQP4+: Aquaporin 4 antibody positive patients, CSF: Cerebrospinal fluid, DN: Double negative patients, LETM: Long segment transverse myelitis, MOGAD: Myelin oligodendrocyte glycoprotein antibody associated disease, MRI: Magnetic resonance imaging, NMOSD: Neuromyelitis optica spectrum disorder, OCB: Oligoclonal band, WM: White matter, *P-value statistically significant
OCT and visual outcome
Twenty two patients (18 with clinical ON and 4 with radiological involvement of optic nerve) underwent detailed ophthalmological assessment, with nine being AQP4+, eight MOGAD and five belonging to DN group.
All had bilateral involvement, thus a total of 44 eyes underwent OCT examination. Half of these patients visited during their first episode of ON, while the rest presented during subsequent relapses. The median number of relapses prior to OCT examination was one (range = 0–7). Patients presenting with the first episode of ON had a significantly thicker RNFL compared to those with recurrent ON (median (range) = 104.5 (50–132) mm vs. 75.5(7–114) mm; P = 0.002). While no significant difference was observed in global RNFL thickness among the three groups, a predilection for nasal RNFL thinning was observed especially in AQP4+ group (P = 0.04) [Supplementary Table 3]. Majority of MOGAD patients showed substantial optic disc swelling apparent on fundoscopy at acute presentation as compared to AQP4+ group (75 % vs. 22.2 %; P = 0.03). Details of OCT and visual outcome parameters in ON patients are shown in [Table 3].
| Parameter | AQP4 (+) (n=18*) A | MOGAD (n=16*) B | DN (n=10*) C | P-value A versus B | P-value A versus B Versus C |
|---|---|---|---|---|---|
| OCT Analysis | |||||
| Median Global RNFL thickness (range)-µm | 94.5 (7–124) | 98.5 (46–132) | 99.5 (61–119) | 0.76 | 0.55 |
| Median Superior quadrant RNFL thickness (range)-µm | 130 (0–154) | 126 (52–164) | 131 (73–143) | 0.73 | 0.57 |
| Median Inferior quadrant RNFL thickness (range)-µm | 123.5 (0–154) | 122 (16–158) | 133 (80–153) | 0.73 | 0.72 |
| Median Nasal quadrant RNFL thickness (range)-µm | 56 (0–102) | 81 (26–106) | 83.5 (56–94) | 0.30 | 0.04 |
| Median Temporal quadrant RNFL thickness (range)-µm | 63 (9–98) | 56 (31–100) | 63 (28–88) | 0.73 | 0.79 |
| Median Macular Volume (range)-µm | 9.2 (6.7–10.1) | 9.2 (6.5–10.1) | 8.6 (8.1–9.7) | 0.98 | 0.52 |
| Median Macular Thickness (range)-µm | 255.5 (198–285) | 255 (226–281) | 248.5 (224–270) | 0.98 | 0.87 |
| VFSS | |||||
| At admission-median (range) | 3 (0–6) | 2.5 (0–6) | 1 (0–3) | 0.65 | 0.12 |
| At discharge-median (range) | 0 (0–6) | 0 (0–3) | 0 (0–0) | NA | NA |
| At 3 months-median (range) | 0 (0–6) | 0 (0–2) | 0 (0–0) | NA | NA |
| At 6 months-median (range) | 0 (0–6) | 0 (0–0) | 0 (0–0) | NA | NA |
| Fundus | |||||
| Optic nerve swelling | 2 (22.2%) | 6 (75%) | 1 (20%) | 0.03 | 0.04 |
DN: double negative patients, MOGAD: Myelin oligodendrocyte glycoprotein antibody associated disease, NMOSD: Neuromyelitis optica spectrum disorder, OCB: oligoclonal band, OCT: optical coherence tomography, RNFL: Retinal nerve fibre layer: VFSS: Visual functional severity scale
Treatment modalities and outcomes
All patients received steroids (Intravenous methylprednisolone [IVMP] 1 g daily for 5 days) during acute episodes. Most of our patients (90.3%) responded to IVMP, with only three requiring rescue therapy, including PLEX in an AQP4+ patient and IVIG in two patients, one each from AQP4+ and DN group. [Table 4] shows the treatment details and outcome in the MOGAD and NMOSD subtypes.
| Parameter | NMOSD whole cohort (n=31) | AQP4+ (n=13) A | MOG+ (n=10) B | DN (n=8) C | P-value A versus B | P-value A versus B versus C |
|---|---|---|---|---|---|---|
| Acute therapy (IVMP) (%) | 31 (100) | 13 (100) | 10 (100) | 8 (100) | ||
| Patients requiring rescue therapy-n% | 3 (9.7) | 2 (15.4) | 0 | 1 (12.5) | - | - |
| Rescue therapy (%) | ||||||
| IVIG | 2 (6.5) | 1 (7.7) | 0 | 1 (12.5) | - | - |
| PLEX | 1 (3.2) | 1 (7.7) | 0 | 0 | - | - |
| Maintenance therapy (%) | ||||||
| Oral steroids alone | 14 (45.2) | 3 (23.1) | 6 (60) | 5 (62.5) | 0.1 | 0.15 |
| Oral steroids+Azathioprine | 12 (38.7) | 7 (53.8) | 3 (30) | 2 (25) | 0.40 | 0.24 |
| Oral steroids+MMF | 3 (9.7) | 3 (23.1) | 0 (0) | 0 (0) | - | - |
| Steroids+Rituximab | 2 (6.5) | 0 (0) | 1 (10) | 1 (12.5) | - | - |
| EDSS-median (range) | ||||||
| At admission | 6 (2–9) | 7 (3–9) | 3.5 (2–9) | 6 (4–9) | 0.01 | 0.02 |
| At discharge | 5.5 (1–9) | 6.5 (2–9) | 2 (1–8) | 6 (2–8) | 0.01 | 0.02 |
| At 3 months | 2 (0–4) | 5.5 (1–8) | 1.5 (0–8) | 5 (0–7) | 0.06 | 0.08 |
| At 6 months | 2 (0–6) | 4.5 (0–7) | 1 (0–10) | 2 (0–5) | 0.16 | 0.27 |
| mRS-median (range) | ||||||
| At admission | 3.5 (2–5) | 4 (2–5) | 2.5 (2–5) | 4 (3–5) | 0.01 | 0.01 |
| At discharge | 3.5 (1–5) | 4 (2–4) | 2 (1–4) | 4 (2–5) | <0.001 | 0.01 |
| At 3 months | 2 (0–4) | 3.5 (0–4) | 2 (0–4) | 3 (0–4) | 0.05 | 0.11 |
| At 6 months | 2 (0–6) | 3 (0–4) | 1 (0–6) | 2 (0–3) | 0.15 | 0.24 |
| Death (%) | 1 (3.6) | 0 | 1 (11.1) | 0 | - | - |
AQP4+: Aquaporin 4 antibody positive patients, DN: Double negative patients, EDSS: Expanded disability status Scale, IVIG: Intravenous immunoglobin G; IVMP: Intravenous methylprednisolone, MMF: Mycophenolate mofetil, MOGAD: Myelin oligodendrocyte glycoprotein antibody associated disease, mRS: Modified Rankin scale, NMOSD: Neuromyelitis optica spectrum disorder, PLEX: Plasma exchange
The EDSS score at admission was significantly better in MOGAD (median [range] = 3.5 [2–9]) as compared to AQP4+ (median [range] = 7.0 [3–9]) and DN (median [range] = 6.0 [4–9]) groups (P = 0.02 for multiple comparisons). Post hoc analysis revealed this significant difference appeared because of differences in admission EDSS scores between AQP4+ and MOGAD groups (P = 0.02). Admission EDSS scores between MOGAD versus DN (P = 0.37) and AQP4+ versus DN groups (P = 0.96) were comparable. One patient each in MOGAD and DN groups was lost to follow-up at 3 and 6 months, respectively. Despite better EDSS scores at admission in the MOGAD group, EDSS scores at 3 (P = 0.08 for multiple comparisons) and 6-month (P = 0.28 for multiple comparisons) follow-up were comparable between the three groups [Supplementary Figure 4]. A good functional outcome (mRS = 0–2) was observed in 56.7% of our patients at 3-month and 62% patients at 6-month follow-up, with MOGAD faring better than DN and AQP4+ patients (80% vs. 71.4% vs. 41.7%; P = 0.15). Despite a negative screening for latent tuberculosis, one of our MOGAD patients who received rituximab, succumbed following an episode of aggressive pulmonary tuberculosis.
DISCUSSION
Nearly three-fourth of our patients with MOGAD and NMOSD subtypes had relapsing course, with TM being the most common clinical presentation followed by ON. As compared to MOGAD, AQP4+ group had female preponderance, frequent dorsal cord LETM, less frequent ON along with preferential involvement of posterior part of optic nerve, including chiasma. Moreover, AQP4+ patients showed a greater nasal RNFL thinning on OCT as compared to MOGAD. DN patients had comparable clinical characteristics to both AQP4+ and MOGAD groups, but a higher frequency of AP syndrome. A good response to corticosteroids was seen in all three groups. While AQP4+ patients had higher admission EDSS score compared to MOGAD, EDSS, and mRS scores at 3 and 6-month follow-up were comparable in the three groups.
MOGAD and NMOSD subtypes accounted for nearly 40% of all demyelinating cases presenting at our center, similar to the previous Indian studies.[9,16] With clarity on NMOSD case definition[17] along with wider availability of immunological tests including serum AQP4 and MOG antibodies, an increase in proportion of these cases in Indian studies was expected.[23] The median (range) age of onset in AQP4+, MOGAD, and DN groups were 39 (18–51), 28.5 (20–61), and 32 (20–55) years, respectively. This was comparable with studies outside India.[24,25] Several Indian studies reported a lower median age of onset, varying between 27 and 33 years, in AQP4+, and 14– 32 years in MOGAD patients,[7,8,13,16,26] likely related to inclusion of pediatric patients in these studies. We studied patients aged >18 years. An Indian study involving patients aged >18 years had age of onset comparable to ours, in all three groups.[6] A strong female preponderance was observed in AQP4+ patients, similar to the previous studies involving different patient populations.[6,7,24,27,28] While a couple of studies reported equal gender prevalence in MOGAD,[16,29] few others reported male predominance[2,24,30] as was seen in our study. Equal gender prevalence in DN patients seen in our study has been reported previously.[2,6] CNS demyelinating disorders including MS and AQP4+ NMOSD have female preponderance.[26,29,31] Equal gender prevalence or male preponderance in MOGAD and DN patients could be related to environmental and/or genetic influences on the study population. Moreover, this supports the speculation that these disorders may have pathophysiology other than CNS demyelination.[29]
TM (38.7%) and ON (22.6%) were the common manifestations at onset, with the former being the most common initial manifestation in all three groups, similar to that reported in other Indian studies.[13,16] Proportion of patients with ON at onset was comparable in all three groups in our cohort, as reported previously.[6] Nearly three-fourth of our patients had relapsing course, similar to previous studies.[6,9,27] Proportion of patients with relapses in AQP4+, MOGAD and DN groups was 85%, 80%, and 54.5%, respectively, in our study, with a couple of Indian studies reporting the same in 92–100%, 60–64%, and 56.5–100%, respectively.[6,13] Majority of relapses in our patients affected the spinal cord (42.6%) and optic nerve (23.5%), with 5.9 % relapses resulting in both TM and ON, which was in line with the previous Indian reports.[6,15,16]
Considering all the 99 demyelinating episodes in our cohort, TM was the most common phenotype in AQP4+ (66%) and DN (64.7%) group, with ON being the commonest in MOGAD (58.6%) patients. TM has been reported as the most common manifestation in AQP4+ NMOSD.[6,7,13,16,26] While some studies reported TM as the commonest manifestation in MOGAD,[6,7,13,16,24] a large Indian study reported ON in nearly half the 263 demyelinating episodes in 93 MOGAD patients.[29] TM was the most common presentation in our DN patients, similar to other reports.[6,13,31] A significantly higher proportion of demyelinating events in MOGAD patients manifested with ON as compared to AQP4+ group, but was comparable to DN group. Increased predilection for ON in MOGAD patients has been previously reported.[7,8,31] Associated features including paroxysmal tonic spasms (PTS) were specifically seen in AQP4+ patients, while headache was reported in all three groups. PTS was seen in 30.8% AQP4+ patients, similar to previous studies.[8,26,31] Although Sato et al. reported PTS in 6.3% of MOGAD and 31.7% of DN patients,[31] no patients in these two groups developed PTS in our study. In accordance with the literature, none of our patient had double antibody positivity.[13,31]
In our study, brain and spinal cord lesions in MRI were seen in 54.8% and 90.3% patients, respectively. MRI brain abnormalities in MOGAD and NMOSD subtypes have been reported in 40–70% of patients.[24,28,31] More than half the patients showed optic nerve lesions, with a significantly higher proportion of AQP4+ patients showing posterior optic nerve and chiasma involvement and MOGAD group having anterior optic nerve (particularly intracanalicular portions) involvement. Involvement of optic chiasma in AQP4+ patients has been more frequently reported as compared to MOGAD by several studies.[6,8,13,16] Ojha et al. 2020 reports significantly higher involvement of anterior segment in MOGAD, as was seen in our study.[8] As compared to AQP4+ and MOGAD, a higher proportion of our DN patients showed MRI brain lesions, with AP and periventricular regions being most commonly involved. Coexisting bilateral ON (25%) along with the presence of LETM (50%) and lack of characteristic lesions of MS in MRI brain in our DN patients argued against the possibility of MS. Although not observed in other Indian studies,[6,13] Sato et al. reported MRI brain lesions in more than half their DN patients, which was comparable to MOGAD but higher than AQP4+ patients.[31] The subgroup of DN patients with a higher brain lesion load may suggest a possibility of yet to be explored neuro-immunological pathway, probably similar to AQP4 receptors, especially in those with predominant AP involvement. More than 80% MOGAD and NMOSD subtypes patients show lesions on MRI of the spinal cord,[13,31] with 90.3% of our patients having the same. In line with the previous reports, majority of our patients had predominant cervicodorsal cord involvement.[8,24,31] LETM involving cervicodorsal cord in AQP4+ patients has been reported,[7,8,31] and was seen in a significantly higher proportion of our AQP4+ patients. Conus medullaris involvement was seen in nearly 30% of our MOGAD patients with cord lesions, comparable with prior studies.[24,31] Cervical cord involvement was also seen in more than two-third of our MOGAD patients, similar to the previous reports.[8,24]
Patients with recurrent ON demonstrated significant RNFL thinning compared to those with the first episode of ON, possibly related to axonal loss and irreversible damage. Involvement of peri-chiasmal optic nerve appears the likely reason for global involvement of RNFL in MOGAD and NMOSD subtypes.[32] Predilection for nasal thinning was seen, especially in the AQP4+ patients. A higher RNFL thinning in AQP4+ patients may result from immune-mediated damage related to abundant AQP4 channels at periphery of retinal peripapillary vessels, which are surrounded by macroglial end foot processes and Müller cells. Immune mediated damage may cause retinal vasculopathy as seen in an observational study,[33] which showed a significantly reduced vessel density in inferior and superior nasal sectors in patients with MOGAD and NMOSD subtypes of CNS disorders. In addition, visual acuity and corresponding VFSS scores improved in a majority of our patients with ON following early treatment with IVMP. One of our AQP4+ patients showed a poor VFSS and significant RNFL thinning at follow-up, probably related to a 24-month delay till first presentation to the hospital.
Acute episodes in most of our patients (90.3%) responded well to IVMP, with only three (9.7%) patients requiring rescue therapy. Earlier guidelines for NMOSD considered azathioprine (with or without prednisolone) and rituximab as first-line therapies.[34] Oral steroids and/or azathioprine was used in 83.9% of our patients. Moreover, the good response to azathioprine observed in our patients, has been reported in previous Indian studies.[9,12,15,16,26] The available evidence, affordability, and easy availability of Azathioprine supports its use as a first-line therapeutic agent for maintenance therapy in Indian patients with these disorders.
EDSS score at 6-month follow-up suggested a higher proportion of our MOGAD patients gaining functional independence compared to AQP4+ and DN groups, although not significant. This could be due to a substantially better baseline functional status of MOGAD patients. Nearly 60% of our patients reported good functional outcome (mRS = 0–2) at 3 and 6-month follow-up. MOGAD patients fared better than DN and AQP4+ patients in our study, which is in line with the previous reports.[8,24,31] While there was no direct disease-related mortality, one of our patients succumbed to aggressive pulmonary tuberculosis following rituximab.
Single-center study and a small sample size are major limitations of this study. However, the prospective nature including follow-up assessment using sufficiently robust outcome parameters are its major strengths. Moreover, this is the first study to compare OCT parameters in an Indian cohort having MOGAD or NMOSD subtypes. A multicenter study with a larger sample size, in clinical settings similar to ours, is desirable to confirm our findings.
CONCLUSION
A relapsing course was seen in nearly three-fourth of patients with MOGAD and NMOSD subtypes, with TM being the most common clinical manifestation. AQP4+ group showed female preponderance, frequent dorsal cord LETM, less frequent ON, and a greater nasal RNFL thinning compared to MOGAD group. Demyelinating lesions on MRI brain were more common in DN patients. All three groups showed good response to pulse steroids in acute phase. EDSS and mRS scores were comparable in the three groups at 3 and 6-month follow-up.
Declaration of patient consent
Institutional Review Board (IRB) permission obtained for the study. The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
SUPPLEMENTARY FIGURES

- Treatment algorithm for NMOSD patients in the study. AQP4+: Aquaporin 4 antibody positive patients, AZA: Azathioprine, CBC: Complete blood count, DN: Double negative patients, EDSS: Expanded disability status scale, IVIG: Intravenous immunoglobin G, IVMP: Intravenous methylprednisolone, LFT: Liver function test, MCV: Mean corpuscular volume, MMF: Mycophenolate mofetil, mRS: modified Rankin scale, NMOSD: Neurom yelitis optica spectrum disorder, RFT: Renal function test, TLC: Total leucocyte count, TPMT: Thiopurine methyltransferase.

- Flow diagram of patient recruitment in the study. AQP4+: Aquaporin 4 antibody positive patients, DN: Double negative patients, EDSS: Expanded disability status scale, MOG-AD: Myelin oligodendrocyte glycoprotein antibody associated disease, mRS: modified Rankin scale, MS: Multiple sclerosis, NMOSD: Neuromyelitis optica spectrum disorder.
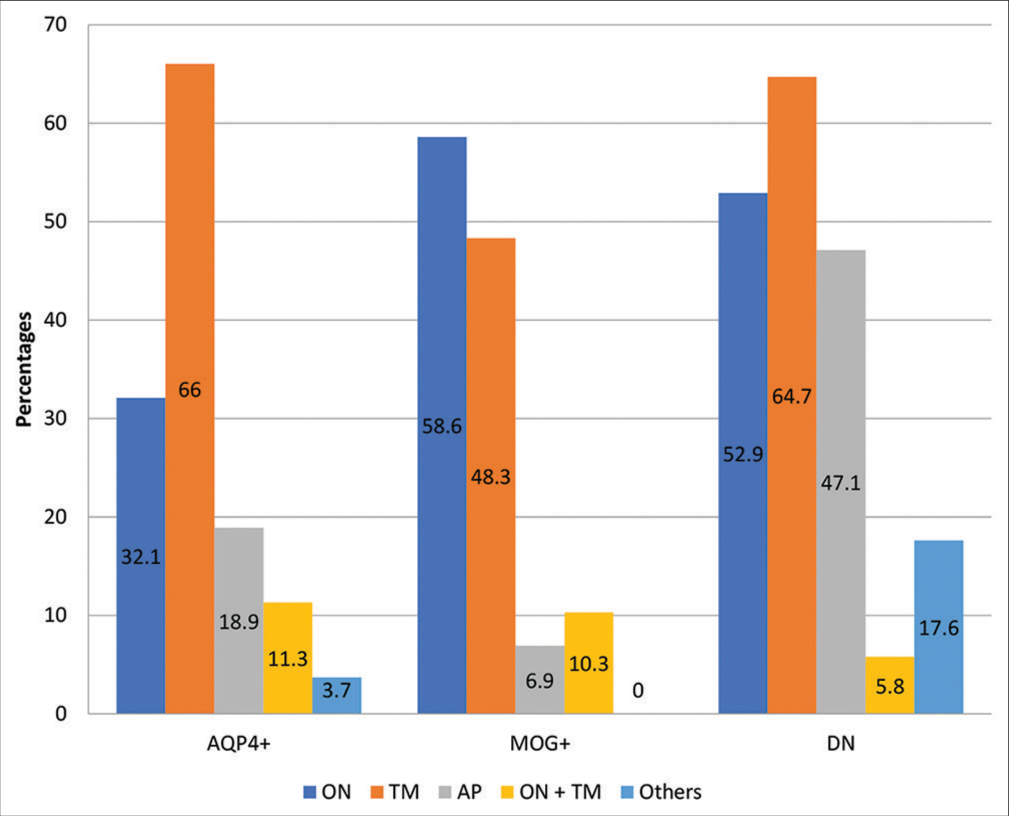
- Proportion of various forms of clinical presentations during the all the demyelinating events in each subtype. AP: Area postrema syndrome, AQP4 +: Aquaporin 4 antibody positive patients, DN: Double negative patients, MOG-AD: Myelin oligodendrocyte glycoprotein antibody associated disease, NMOSD: Neuromyelitis optica spectrum disorder, ON: Optic neuritis, ON+TM: Optic neuritis with transverse myelitis, TM: Transverse myelitis. *Others include involvement of multiple neuraxis including brainstem, diencephalon and cerebral cortex.
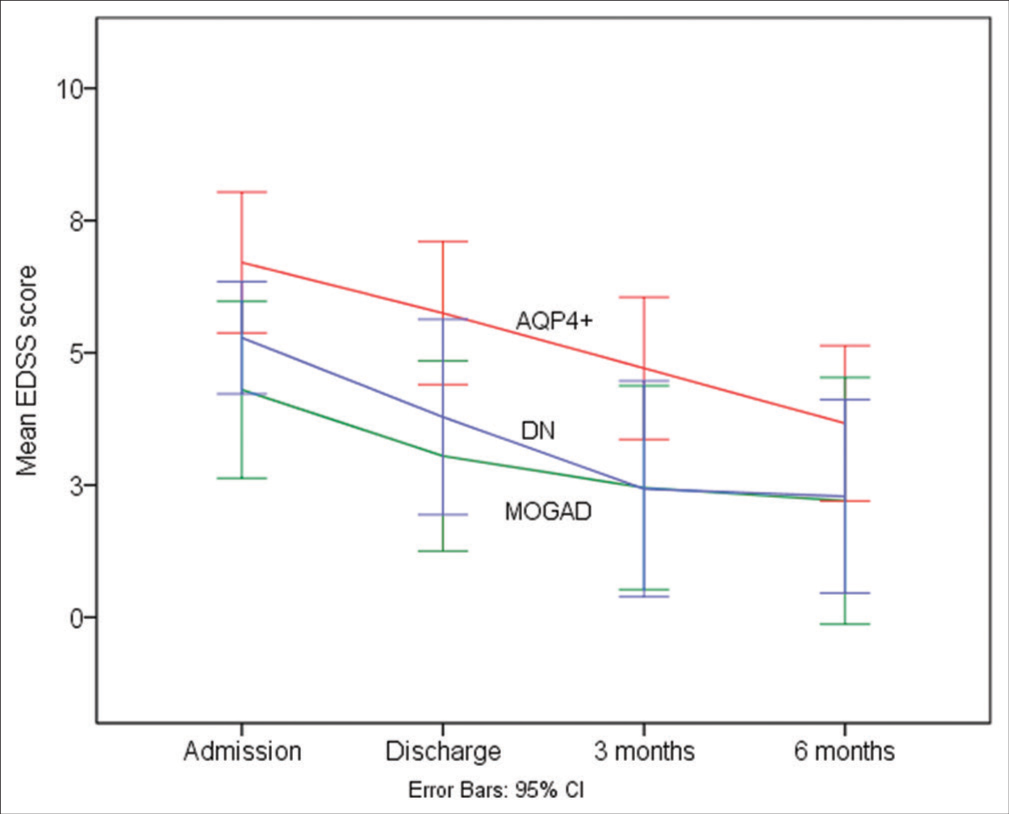
- Serial median EDSS score assessment (admission, discharge, 3 months and 6 months) in AQP4 positive, MOG-AD and Double negative groups. AQP4+: Aquaporin 4 antibody positive patients, DN: Double negative patients, EDSS: Expanded disability status scale, MOG-AD: Myelin oligodendrocyte glycoprotein antibody associated disease, NMOSD: Neuromyelitis optica spectrum disorder.
SUPPLEMENTARY TABLES
| 0 | Normal |
| 1 | Scotoma with visual acuity (corrected) better than 20/30 |
| 2 | Worse eye with scotoma with maximal visual acuity (corrected) of 20/30–20/59 |
| 3 | Worse eye with large scotoma, or moderate decrease in fields, but with maximal visual acuity (corrected) of 20/60–20/99 |
| 4 | Worse eye with marked decrease of fields and maximal visual acuity (corrected) of 20/100–20/200; grade 3 plus maximal acuity of better eye of 20/60 or less |
| 5 | Worse eye with maximal visual acuity (corrected) less than 20/200; grade 4 plus maximal acuity of better eye of 20/60 or less |
| 6 | Grade 5 plus maximal visual acuity of better eye of 20/60 or less |
| Clinical features | NMOSD (n=68) | AQP4+(n=40) A | MOG-AD (n=19) B | DN (n=9) C | P-value A versus B | P-value A versus B versus C | |
|---|---|---|---|---|---|---|---|
| 1 | Isolated ON: n(%) | 16 (23.5) | 6 (15.0) | 8 (42.1) | 2 (22.2) | 0.04* | 0.07* |
| 2 | Isolated TM: n(%) | 29 (42.6) | 19 (47.5) | 6 (31.6) | 4 (44.4) | 0.25 | 0.51 |
| 3 | Isolated AP: n(%) | 5 (7.4) | 3 (7.5) | 1 (5.3) | 1 (11.1) | >0.99 | 0.86 |
| 4 | ON+TM: n(%) | 4 (5.9) | 3 (7.5) | 1 (5.3) | 0 (0.0) | >0.99 | 0.68 |
| 5 | Others*: n(%) | 14 (20.6) | 9 (22.5) | 3 (15.8) | 2 (22.2) | 0.73 | 0.83 |
| Parameter | Relapsing on (n=22*) A | First episode on (n=22*) B | P-value A versus B |
|---|---|---|---|
| Median RNFL thickness (range)- µm | 75.5 (7–114) | 104.5 (50–132) | 0.002* |
| Median macular volume (range)- µm | 8.9 (6.7–10.3) | 9.25 (6.5–10.1) | 0.450 |
| Median macular thickness (range)- µm | 247 (198–285) | 258 (226–285) | 0.226 |
Financial support and sponsorship
Nil.
References
- Neuromyelitis optica spectrum disorder and other non-multiple sclerosis central nervous system inflammatory diseases. Contin Lifelong Learn Neurol. 2019;25:815-44.
- [CrossRef] [PubMed] [Google Scholar]
- Prevalence and patterns of demyelinating central nervous system disorders in urban Mangalore, South India. Mult Scler J. 2014;20:1651-3.
- [CrossRef] [PubMed] [Google Scholar]
- What proportion of AQP4-IgG-negative NMO spectrum disorder patients are MOG-IgG positive? A cross sectional study of 132 patients. J Neurol. 2017;264:2088-94.
- [CrossRef] [PubMed] [Google Scholar]
- Optical coherence tomography helps differentiate neuromyelitis optica and MS optic neuropathies. Neurology. 2009;73:302-8.
- [CrossRef] [PubMed] [Google Scholar]
- A clinical and radiological profile of neuromyelitis optica and spectrum disorders in an Indian cohort. Ann Indian Acad Neurol. 2014;17:77-9.
- [CrossRef] [PubMed] [Google Scholar]
- A comparative analysis of clinical and imaging features of Aquaporin 4 (AQP4) antibody positive, Myelin oligodendrocyte glycoprotein (MOG) antibody positive and double seronegative subtypes of neuro myelitis optica spectrum disorder (NMOSD) Ann Indian Acad Neurol. 2022;25:239-45.
- [CrossRef] [PubMed] [Google Scholar]
- Comparison of clinical and radiological features of Aquaporin4 (AQP-4) antibody positive neuromyelitis optica spectrum disorder (NMOSD) and anti myelin oligodendrocyte glycoprotein (Anti-MOG) syndrome-our experience from Northwest India. Ann Indian Acad Neurol. 2022;25:246-55.
- [CrossRef] [PubMed] [Google Scholar]
- Myelin oligodendrocyte glycoprotein (MOG) antibody-associated CNS demyelination: Clinical spectrum and comparison with aquaporin-4 antibody positive neuromyelitis optica spectrum disorder. Neurol India. 2020;68:1106-14.
- [Google Scholar]
- Anti-myelin oligodendrocyte glycoprotein antibody associated disease spectrum-A north Indian tertiary care centre experience and review of literature. J Neuroimmunol. 2020;340:577143.
- [CrossRef] [PubMed] [Google Scholar]
- Myelin oligodendrocyte glycoprotein (MOG)-IgG associated demyelinating disease: Our experience with this distinct syndrome. Ann Indian Acad Neurol. 2021;24:69-77.
- [CrossRef] [PubMed] [Google Scholar]
- Neuromyelitis optica spectrum disorders: An experience from tertiary care hospital of North-West India. Indian J Med Spec. 2017;8:192-6.
- [CrossRef] [Google Scholar]
- Clinical features, gender differences, disease course, and outcome in neuromyelitis optica spectrum disorder. Ann Indian Acad Neurol. 2021;24:186-91.
- [CrossRef] [PubMed] [Google Scholar]
- Serological markers associated with neuromyelitis optica spectrum disorders in South India. Ann Indian Acad Neurol. 2016;19:505-9.
- [CrossRef] [PubMed] [Google Scholar]
- Neuromyelitis optica spectrum disorders in North Indian population: Experience from a tertiary care center. Neurol India. 2022;70:1500-5.
- [CrossRef] [PubMed] [Google Scholar]
- Neuromyelitis optica and neuromyelitis optica spectrum disorder: Natural history and long-term outcome, an Indian experience. J Neurosci Rural Pract. 2015;6:331-5.
- [CrossRef] [PubMed] [Google Scholar]
- Clinicoradiological comparative study of Aquaporin-4-IgG seropositive neuromyelitis optica spectrum disorder (NMOSD) and MOG antibody associated disease (MOGAD): A prospective observational study and review of literature. J Neuroimmunol. 2021;361:4-9.
- [CrossRef] [PubMed] [Google Scholar]
- International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. 2015;85:177-89.
- [CrossRef] [PubMed] [Google Scholar]
- Association of MOG-IgG serostatus with relapse after acute disseminated encephalomyelitis and proposed diagnostic criteria for MOG-IgG-associated disorders. JAMA Neurol. 2018;75:1355-63.
- [CrossRef] [PubMed] [Google Scholar]
- Visual acuity changes in patients with leber congenital amaurosis and mutations in CEP290. JAMA Ophthalmol. 2013;131:178.
- [CrossRef] [PubMed] [Google Scholar]
- Improving the assessment of outcomes in stroke: Use of a structured interview to assign grades on the modified Rankin Scale. Stroke. 2002;33:2243-6.
- [CrossRef] [PubMed] [Google Scholar]
- Rating neurologic impairment in multiple sclerosis: An expanded disability status scale (EDSS) Neurology. 1983;33:1444-52.
- [CrossRef] [PubMed] [Google Scholar]
- Therapeutic options in neuromyelitis optica spectrum disorders. Expert Rev Neurother. 2016;16:319-29.
- [CrossRef] [PubMed] [Google Scholar]
- Neuromyelitis optica spectrum disorders with aquaporin-4 and myelin-oligodendrocyte glycoprotein antibodies a comparative study. JAMA Neurol. 2014;71:276-83.
- [CrossRef] [PubMed] [Google Scholar]
- Antibodies to MOG and AQP4 in adults with neuromyelitis optica and suspected limited forms of the disease. Mult Scler J. 2015;21:866-74.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical, neuroimaging and therapeutic response in AQP4-positive NMO patients from India. Mult Scler Relat Disord. 2019;30:85-93.
- [CrossRef] [PubMed] [Google Scholar]
- Neuromyelitis optica in France: A multicenter study of 125 patients. Neurology. 2010;74:736-42.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical features of neuromyelitis optica in a large Japanese cohort: Comparison between phenotypes. J Neurol Neurosurg Psychiatry. 2011;82:1360-4.
- [CrossRef] [PubMed] [Google Scholar]
- Myelin oligodendrocyte glycoprotein-antibody-associated disorder: A new inflammatory CNS demyelinating disorder. J Neurol. 2021;268:1419-33.
- [CrossRef] [PubMed] [Google Scholar]
- Neuromyelitis optica spectrum disorders without and with autoimmune diseases. BMC Neurol. 2014;14:162.
- [CrossRef] [Google Scholar]
- Distinction between MOG antibody-positive and AQP4 antibody-positive NMO spectrum disorders. Neurology. 2014;82:474-81.
- [CrossRef] [PubMed] [Google Scholar]
- Role of optical coherence tomography in determining specific diagnostic pattern of retinal nerve fiber layer involvement in various central nervous system demyelinating diseases: An observational study. Neurol Clin Neurosci. 2021;9:376-80.
- [CrossRef] [Google Scholar]
- Optical coherence tomography angiography of peripapillary vessel density in multiple sclerosis and neuromyelitis optica spectrum disorder: A comparative study. J Clin Med. 2021;10:609.
- [CrossRef] [PubMed] [Google Scholar]
- EFNS guidelines on diagnosis and management of neuromyelitis optica. Eur J Neurol. 2010;17:1019-32.
- [CrossRef] [PubMed] [Google Scholar]




