
Translate this page into:
Alkaptonuria, more than just a mere disease
Address for correspondence: Dr. Ismael Abdullah, Parkinson's Clinic of Eastern Toronto and Movement Disorders Center, 1-2060 Ellesmere Road, Toronto, ON, Canada - M1H 2V6. E-mail: ismaelabdullah@hotmail.com
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Alkaptonuria (AKU) is considered a rare autosomal recessive condition that results in an accumulation of homogentisic acid in body tissues and causes long-term clinical, neurological and psychological complications. We present a comprehensive evaluation of an atypical 46-year-old Caucasian male who developed all clinical diagnostic symptoms of AKU (ochronotic pigmentations, dark urine and clinical arthritis of major joints including spine) by 25 years of age, well before the typical age mentioned in many reviews. First signs of ochronotic ear pigmentations unexpectedly started appearing as early as 12 years of age. A long “disease-free” period typical in classical AKU patient was also absent. This case report highlights the importance of considering psychological issues in AKU patients. The patient showed symptoms of dysthymia reporting social isolation, diminished interest in pleasurable activities, feeling of worthlessness and irritability as major psychological issues. Early ochronotic pigmentation, advanced spinal myelopathy and arthropathy of major joints suggests aggressive course of the disease. Our patient underwent bilateral shoulder replacement due to AKU-induced arthropathy resulting in restoration of some range of motions. AKU is not fully understood and we recommend treating it as a multidimensional disease with simultaneous physiological, neurological and psychological effects. Early diagnosis, understanding of disease prognosis and emphasis on psychological health is needed to improve the quality of life of AKU patients.
Keywords
Alkaptonuria
arthropathy
depression
myelopathy
ochronosis

Introduction
Termed as an “inborn error of metabolism” by Sir Archibald Garrod,[1] alkaptonuria (AKU) is often considered an autosomal recessive condition[2] with an estimated frequency ranging from 1:250,000 live births in the USA[3] to 1:19,000 in Slovakia.[4] Inheritance of AKU is not fully clear (uncommon cases of dominant inheritance have been reported before in AKU families).[2] Genetically, AKU results from mutations in a 54,363 bp gene (mapped on chromosome 3q) that codes for an enzyme homogentisate 1,2-dioxygenase (HGD).[3] HGD is a key metabolic enzyme responsible for converting homogentisic acid (HGA) to maleylacetoacetic acid during phenylalanine and tyrosine catabolism.[5] Metabolically, non-functional HGD enzyme results in the accumulation of HGA in the body despite normal renal clearance. Excess HGA undergoes oxidative conversion in tissues (as in standing urine) into melanin-like polymers.[6] Accumulation of HGA and its metabolites in tissues results in ochronosis, a term used to describe the darkening of tissues.[6] Symptoms of AKU include ear and scleral pigmentation, arthropathy of major joints, calcification of cartilaginous tissue as well as deterioration of cardiac valves.[36] Current treatment focus is on symptomatic care and may include administering analgesics, ascorbic acid, diet restrictions or surveillance.[6] As disease progresses, invasive procedures such as joint surgeries or organ transplantations may be recommended. Physiotherapy and lifestyle counseling have been underutilized and no study exists to evaluate their effectiveness. Psychological effects of AKU have also not been recognized.
Our case study presents atypical aggressive progression of AKU with focus on physiological, psychological and neurological effects that are underreported or not reported before, to the best of our knowledge.
Case Report
A 46-year-old Caucasian male was referred to our neurology clinic due to sensory symptoms of lower extremities. Presence of dark urine at 8 years of age revealed that he was diagnosed with AKU (urinalysis and biochemical testing confirmed the presence of HGA at the time of diagnosis). He developed progressive lower back and shoulder pain before 25 years of age and first signs of ochronosis were evident as early as 12 years of age on pinna of ears. Family history was unremarkable (no report of osteoarthritis, AKU or consanguinity). The patient had extensive arthritic and cartilage damage on all weight bearing joints of knees, hips and vertebral column. Shoulder joints were the most severely affected showing extensive swelling, limited mobility and arthritic degeneration [Figures 1 and 2]. Clinical examination of the spine showed impaired cervical, thoracic and lumbar mobility. An MRI of spine displayed multi-level disc space narrowing, spinal cord compression and disc herniations at cervical and lumbar spine with effacement of cerebrospinal fluid space [Figures 3 and 4]. The patient experienced persistent tingling, diffuse constant pain and numbness in both of his feet and toes. Motor testing of the lower extremities showed normal muscle tone and strength, however, motor movements of the upper extremities were sluggish and painful, and muscle strength was slightly weak. Sensory testing of the extremities to pin prick, light, touch, vibration and temperature was normal. Examination of the patient's gait was also normal. Patient reported no trouble with vision, speech or swallowing. Due to extensive shoulder arthropathy and cartilage damage, the patient required bilateral shoulder replacement and underwent total right shoulder replacement surgery. The patient showed symptoms of dysthymia, although he did not meet formal criteria for depression. Limited mobility, decreased range of motion (ROM) and lost function of day-to-day basic activities initiated irritability, social isolation, feeling of worthlessness and diminished interest in pleasurable activities as major psychological issues. The surgery improved some ROM. The patient adhered to a low protein diet. The patient was administered multiple steroid injections and analgesics in both shoulders over the past 3 years to manage pain. Despite some reduction in pain, the injections were not very effective to change the quality of life. Our study is in contrast to other findings that report no correlation between disability of AKU patients with severe impairments and self-reports of mental competencies.[7]

- CT scan of left (left image) and right (right image) shoulder showing advance arthropathy of glenohumeral joint with total loss of joint space, marked subchondral sclerosis and subchondral cyst formation
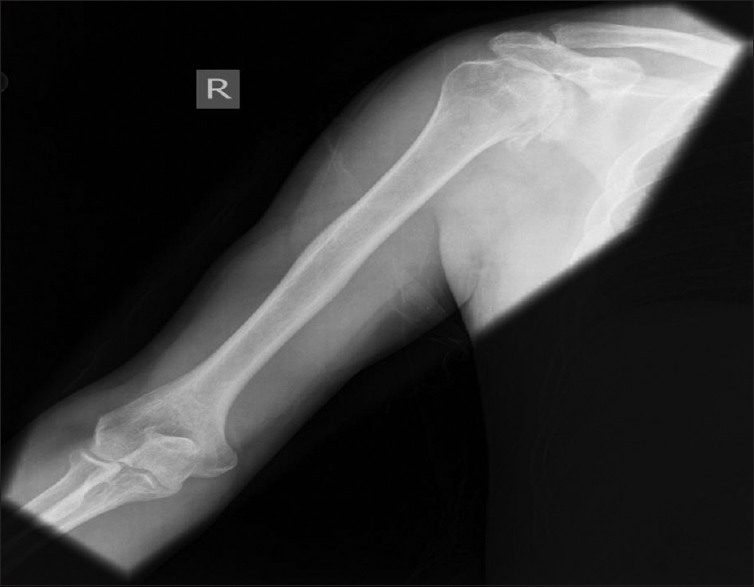
- An X-ray of right shoulder showing markedly advanced degenerative change with substantial narrowing of the glenohumeral joint space, significant sclerosis and subchondral cyst formation
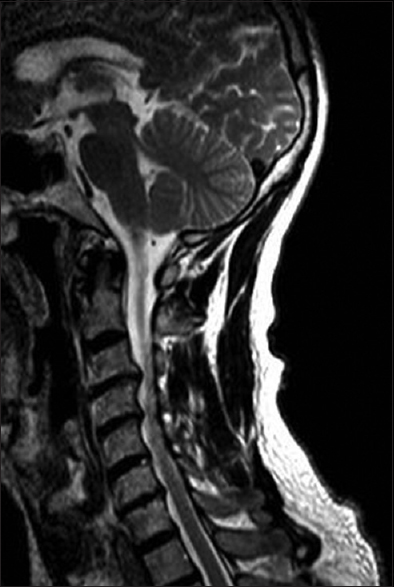
- A sagittal view of T2-weighted MRI of C-spine displayed multi level moderate disc space narrowing, diffuse disc bulges, osteophyte formation, and disc herniations at C3-C4, C4-C5 with effacement of cerebrospinal fluid space, mass effect on spinal cord with spinal cord compression and abnormal T2 signal changes in spinal cord at these levels
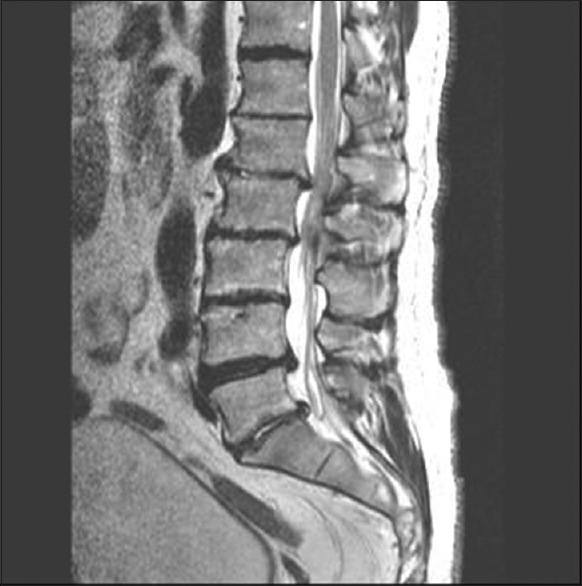
- A sagittal view of T2-weighted MRI of lumbar spine showing moderate degenerative disc disease at L1-2, L2-3, L4-5, L5-S1
Discussion
Reviews indicate that physical symptoms of AKU, particularly ochronotic ear and scleral pigmentations, almost always appear after 30 years of age with progressive arthritic and organ changes as age advances.[36] Our patient unexpectedly developed ear ochronosis at 12 years of age, much earlier than typical presentations. In fact, tetrad of physical diagnostic features of AKU: (i) Dark urine, (ii) Black scleral pigmentation, (iii) Black/blue ear pigmentation and (iv) Clinical arthritis (with lower back pains), were all present in this case as early as 25 years of age. AKU patients are asymptomatic as children or young adults.[8] A long “disease-free” period—no physical symptoms until 28-30 years of age except dark upon standing—typical in classical AKU patient was absent in this case. Such progressions of AKU are rarely reported. The youngest patient with similar symptoms of AKU (excessive HGA in urine, ochronosis and arthropathy of major joints) reported until now was 14 years old.[9] Early manifestations of AKU, such as dark urine are usually noticed by mothers when changing the diaper of a newborn.[10] Adults usually complain of a dark brown to black staining on their clothing near the axilla.[10] Other secondary complications experienced by our patient included persistent tingling and numbness on lower extremities (thigh, toes and feet) maybe caused by significant degenerative changes at all levels of spinal cord, and resulted in disruption of normal transmission of spinal signals. Despite the high prevalence of intervertebral disc degeneration in AKU patients, neurologic symptoms caused by spinal diseases are rare.[11] Spinal compression, disc bulges, herniations as well as effacement of cerebrospinal fluid space abnormally affect the spinal signals resulting in multitude of symptoms ranging from numbness, tingling, and bladder problems to diffuse pain. Hence, numbness and tingling in lower extremities are indicative of advanced arthritic changes in spine. Investigations on disc protrusions are often neglected since most association with AKU is pain in the lower back and legs.[12] Surgical procedures to treat spinal cord compressions in AKU patients have shown significant neurological improvements.[12]
This case highlights the importance of psychological factors in AKU patients. Due to lifelong progressive nature of the disease and resulting disability, patients often develop psychological issues, which are largely ignored by medical practitioners. Quality of life of AKU patients can best be improved by managing both physiological and psychological health. Our patient received counseling from his family physician and neurologist with focus on diet and life style modifications. With some success, the severity of depression, social isolation and irritability decreased. After bilateral shoulder replacement surgery, the patient reported better success in ROM and more participation in basic day-to-day activity. Both counseling and surgery improved the physiological and psychological health of our patient.
This case report is noteworthy due to the aggressive nature of the disease that include early ochronotic pigmentation (<20 age), advanced spinal compression and myelopathy, and arthropathy of major joints. In a recent publication, an 80-year-old woman of Austrian descent was diagnosed with ochronosis of hip joints caused by the accumulation of HGA in connective tissues (including hyperpigmentation of ear and scleral tissue).[13] Our patient was presented with similar symptoms at a much younger age, including shoulder immobility due to AKU induced arthropathy that required bilateral surgery, not previously reported to the best of our knowledge.
Currently, no effective treatment for AKU is known and focus is on symptomatic care. Rarity of a progressive disease with no cure, like AKU, calls for a better understanding of diagnosing the disease. Recent trials with nitisinone (drug that blocks HGA production) showed that it effectively decrease HGA levels, but results in increased plasma tyrosine.[14] Pathophysiological and clinical significance of tyrosinemia are unknown.[3] Our patient underwent right shoulder replacement surgery that decreased shoulder pain and restored some ROMs. Lifestyle counseling that includes diet control, strategies to manage pain and focus on hobbies/activities that can minimize loading of major joints can also benefit patients. In addition to lifestyle counseling, psychotherapy may also be helpful and can allow a patient to talk about the disability and pain while feeling supported. Lifestyle counseling and psychotherapy effectiveness in treating alkaptonuria is unknown. We recommend more studies to evaluate its value.
Conclusions
AKU has never been fully understood and we recommend treating alkaptonuria as a multidimensional disease with progressive physiological, neurological and psychological effects. Early diagnosis, understanding of disease prognosis and emphasis on mental health could improve the quality of life of patients affected by this rare disease.
Source of Support: Nil.
Conflict of Interest: None declared.
References
- Garrod's Croonian Lectures (1908) and the charter ‘Inborn Errors of Metabolism’: Albinism, alkaptonuria, cystinuria, and pentosuria at age 100 in 2008. J Inherit Metab Dis. 2008;31:580-98.
- [Google Scholar]
- Three-generational alkaptonuria in a non-consanguineous family. J Inherit Metab Dis. 2008;31(Suppl 2):S425-30.
- [Google Scholar]
- Recent advances in management of alkaptonuria (invited review; best practice article) J Clin Pathol. 2013;66:367-73.
- [Google Scholar]
- High frequency of alkaptonuria in Slovakia: Evidence for the appearance of multiple mutations in HGO involving different mutational hot spots. Am J Hum Genet. 2000;67:1333-9.
- [Google Scholar]
- The nature of the defect in tyrosine metabolism in alcaptonuria. J Biol Chem. 1958;230:251-60.
- [Google Scholar]
- Musculoskeletal findings and disability in alkaptonuria. J Rheumatol. 2006;33:2280-5.
- [Google Scholar]
- Nine cases of Alkaptonuria in one family in southern Jordan. 2012. Rheumatol Int. 2012;32:621-5.
- [Google Scholar]
- Early-onset ocular ochronosis in a girl with alkaptonuria (AKU) and a novel mutation in homogentisate 1, 2-dioxygenase (HGD). Prilozi-Maked. Akad. Nauk. Umet., Odd. Biol. Med. Nauki. 2011;32:305-11.
- [Google Scholar]
- Alkaptonuric ochronosis presenting as palmoplantar pigmentation. Clin Exp Dermatol. 2000;25:305-7.
- [Google Scholar]
- Alcaptonuria with lumbar disc prolapse: Case study and review of the literature. Spine J. 2007;7:495-8.
- [Google Scholar]
- Australian Government. Australian Public Assessment Report for Nitisinone. Australia: Department of Health and Aging; 2011. p. :1-86.
- [Google Scholar]




