
Translate this page into:
A study of Guillain–Barré syndrome with reference to cranial neuropathy and its prognostic implication
Address for correspondence: Dr. Basavaraj F. Banakar, Department of Neurology, Mahatma Gandhi Hospital, Jodhpur - 342 001, Rajasthan, India. E-mail: drbanakar2006@yahoo.co.in
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Background:
Focused studies on cranial neuropathy in Guillain–Barré syndrome (GBS) and its prognostic implication are not done previously.
Aim:
To study the clinical profile of GBS patients with special reference to cranial neuropathy and its prognostic implication.
Materials and Methods:
The study included 61 patients with GB syndrome, fulfilling Asbury Cornblath's criteria for GB syndrome. A pre-designed semi-structured questionnaire was used to obtain data regarding demographic profile and clinical profile. All patients underwent detailed neurological examination, investigations including nerve conduction studies and CSF examination and treated according to the severity of the illness. Patients were followed up for 6 months. During analysis two groups were made depending on cranial nerve involvement, and compared with respect to various parameters.
Results:
Out of 61 patients 38 (62.3%) patients had cranial nerve palsies, in that 25 had multiple cranial nerve palsies, and 13 had single isolated nerve palsy. A majority of 30 (49.2%) had bulbar palsy, 28 (46%) had facial nerve palsy, and all had bilateral involvement except 3 patients who had unilateral palsy. Hypoglossal nerve involvement was seen in six (10%) patients and four (6.5%) patients had ophthalmoplegia. Only one had bilateral vestibulocochlear nerve palsy. On comparing various clinico-electrophysiological parameters among patients of GB syndrome with and without cranial nerve involvement, the presence of respiratory paralysis, IVIg and ventilatory support requirement had significant association with cranial nerve involvement in GBS.
Conclusion:
Our study found a correlation between cranial nerve palsies and severity of the illness. Cranial nerve innervated muscles recover earlier as compared to distal limb muscles. No association was found between outcome at 6 months and cranial nerve involvement.
Keywords
Bulbar palsy
facial palsy
Guillain–Barré syndrome

Introduction
Guillain–Barré syndrome (GBS) is an acute inflammatory polyradiculoneuropathy, characterized by a rapidly progressive, symmetric, areflexic flaccid paralysis with albuminocytological dissociation.[1] The incidence of typical GBS varies between 0.6 and 4 cases per 100,000 per year throughout the world.[2] With recent eradication of poliomyelitis, the GBS is currently the most frequent cause of acute flaccid paralysis in India and constitutes one of the serious emergencies in neurology. It can affect all ages, with male predominance including children. GBS has been frequently associated with preceding nonspecific infection or triggering factors like trauma, surgery or vaccination usually a few days to weeks before the onset of neurological symptoms.
The diagnosis of GBS is clinical but may be aided by electrophysiology which is also important to characterize the subtypes: Acute inflammatory demyelinating polyradiculoneuropathy (AIDP), acute motor axonal neuropathy (AMAN), and acute motor and sensory axonal neuropathy (AMSAN).[3] GBS patients often develop cranial nerve weakness, usually in the form of facial or pharyngeal weakness.[4] There are no studies in the literature specifically focused on cranial nerve palsies in GBS patients. Hence, we took up this study along with clinical profile of GBS patients.
Materials and Methods
All patients with GBS fulfilling the Asbury Cornblath criteria, admitted or referred to us between 1st Oct 2011 and 31st Dec 2013 were included. The study was approved by the Institutional Ethics Committee. The demographic profile including age, sex, occupation, month of illness, preceding antecedent illness, and duration of the disease were recorded. Detailed clinical history and neurological examinations were done. Patient's disability at the peak of deficit was assessed using Hughes[5] functional grading [Table 1], up to second grade taken as good and above that as bad score. Special emphasis was given to the cranial nerve involvement, with daily enquiry and examination for dysphagia, facial asymmetry, diplopia and dysarthria. All the patients underwent investigations including complete hemogram, serum biochemistry, potassium, serology for anti-nuclear antibody (ANA), hepatitis B surface antigen (HBsAg), and human immunodeficiency virus (HIV), urine for protein, CSF studies and detailed nerve conduction studies (NCS).

NCSs were performed within second week of hospitalization. Study included median, ulnar, common peroneal, tibial and sural nerves using conventional techniques. In our study, the patients were classified into AIDP, AMAN and AMSAN based on Ho, et al.[6] criteria. Depending on the severity of the illness patients were treated. Intravenous Immunoglobulin (IVIg) was used as immunotherapy, at a dose of 2 g/kg body weight in divided doses over 5 days, for patients having the indications. All the patients were followed up at regular monthly intervals. Recovery and disability were recorded using functional grading scale of Hughes, et al.[5] Detailed enquiry and examination were done for any residual cranial nerve deficit at every visit.
During analysis two groups were made depending on cranial nerve involvement:
-
Cranial nerve group (CN) and
-
Non-cranial nerve group (NCN).
These groups were compared with reference to various parameters including demographic profile, sensory, autonomic and respiratory involvement, requirement of ventilator support and IVIg, duration of hospital stay, Hughes score at admission and during follow up.
Statistical analysis
A database was created in MS Excel and after appropriate cleaning; analysis was performed using SPSS Version 16. Appropriate descriptive statistics like proportion and percentage were done. Appropriate tests were used to analyze these data.
Results
The study comprises of 61 GBS patients in which 44 (72.1%) were males, 17 (27.9%) were females, and majority (78%) were from rural background. The age group ranged from 8 to 78 years (mean age 34 years) with majority being in the age group of 11 to 40 years. There was seasonal variation in the incidence of the disease, highest being during Aug-Nov (44.3%).
Of the 61 patients antecedent events were observed in 29 (47.5%) and included upper respiratory illness in 12 (41.3%), acute gastroenteritis in 10 (34.4%) and only fever in 7 (24.1%) patients. The latent period between the illnesses ranged from 2 to 20 days. Most common presenting symptom was motor weakness in 98% of patients. Around 70% (43) had subjective sensory complaints, in them 22 (36.1%) had limb pain, 13 (21.3%) had paresthesia in limbs, 5 (8.2%) had backache and 3 (5.9%) combined limb/back pain, but objectively only 6 (10%) had evidence of sensory loss. Respiratory involvement was present in 31 (51%) of patients. Autonomic dysfunction was seen in 27 (44.3%) patients which included tachycardia (10), constipation (7), bradycardia (4), excessive sweating (2) and hypertension (2). Bladder involvement was seen in 9 (14.8%) patients.
Atypical features in the form of seizures were seen in three patients. Seizures were secondary to progressive reversible encephalopathy syndrome (PRES) in two patients and in one following hypoxic damage in the brain. Cause for PRES was dysautonomia in one patient and following IVIg therapy in the other.
Different subtypes of GBS based on electrophysiological abnormalities included AIDP in 37 (60.6%), AMAN 20 (32.8%) and AMSAN in 3 (5%) according to Ho, et al. criteria. In one patient it was indeterminate. Cerebrospinal fluid analysis was showing albuminocytological dissociation in 49 (80%) patients, in which protein level ranged from 48 to 242 mg/dl. Majority (95%) patients had bad Hughe's score at the nadir of illness. At our center we don’t have facilities for plasmapheresis hence IVIg is the only immunotherapy, and it was given in 42 (69%) patients. Ventilator support was given in 22 (36%) patients. Duration of ventilator support ranged from 2 to 45 days, two patients died. Total duration of hospital stay ranged from 4 to 65 days. All these patients were followed every monthly, disability recorded according to Hughes score. At the end of 1 month only 4 (6.5%) patients had good outcome score [Hughes ≤ 2], remaining 55 (90%) had poor outcome score [Hughes ≥ 3]. But at the end of 6 months, 8 patients lost to follow up, in the remaining 38 (74.5%) had good outcome score, i.e. they were able to walk unassisted and 13 (25.5%) had bad outcome score. The most common residual sign at the end of 6months was the absence of ankle jerk which was seen in 25 patients.
Cranial neuropathy
62.3% (38) of patients had cranial nerve palsies, in that 25 had multiple cranial nerve palsies, and 13 had single type of nerve palsy [Table 2]. In single type of nerve involvement seven had facial palsy and six had bulbar palsy. Overall, as shown in figure 1 majority had bulbar palsy, 30 (49.2%). Facial nerve palsy was seen in 28 (46%), all had bilateral involvement except 3 patients who had unilateral palsy. Hypoglossal nerve involvement was seen in six (10%) patients and four (6.5%) patients had ophthalmoplegia. Only one had vestibulocochlear nerve palsy. All these were followed up at monthly interval, at the end of 3 months all had complete recovery of cranial palsies, facial nerve being last to recover.
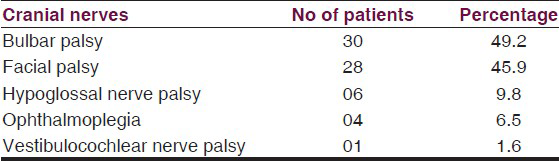
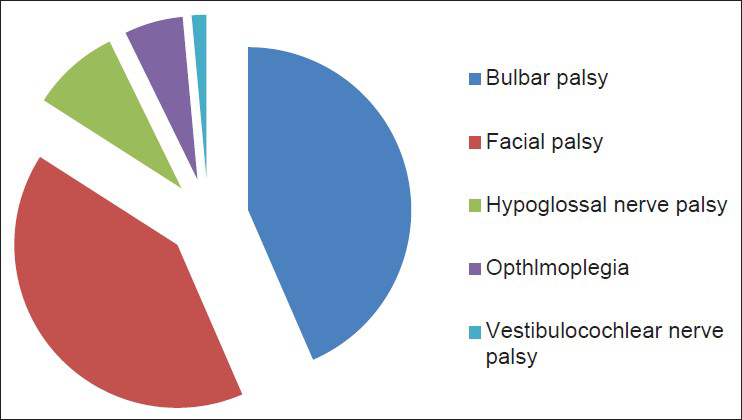
- Pie diagram showing percentage involvement of cranial nerves
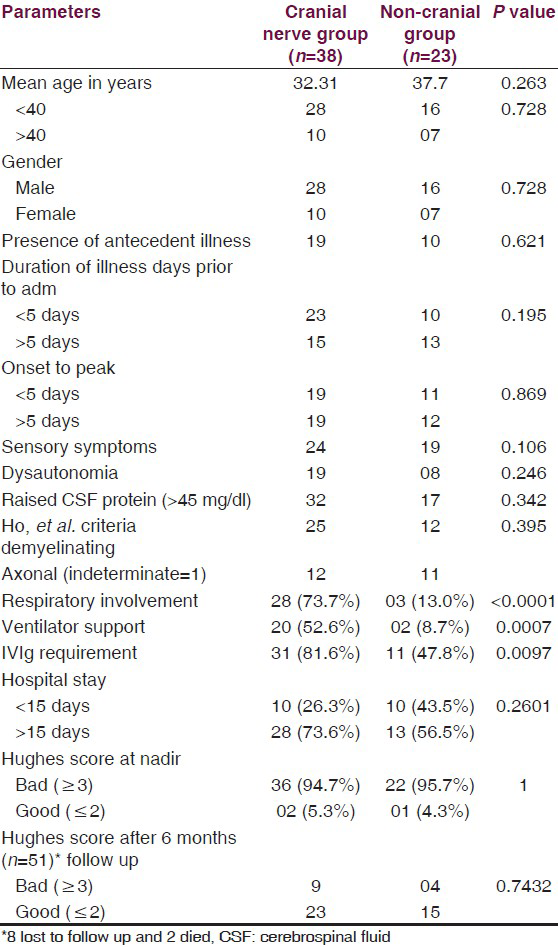
On comparing various parameters among patients of GBS with and without cranial nerve involvement [Table 3], the statistically significant difference was seen with respect to respiratory involvement, IVIg requirement and ventilator support.
Discussion
The Guillain–Barré syndrome (GBS) is an acute-onset, monophasic, immune-mediated polyneuropathy that often follows an antecedent infection.
The first description of what is now called the Guillain–Barré syndrome, was given by J.B.O. Landry in 1859.[7] The summation of clinical characteristics and typical findings in the cerebrospinal fluid as described by Guillain, Barre and Strohl in 1916.[8] As research progressed, many studies reported on the clinical diversity of GBS. As a consequence, the concept of GBS shifted from a single clinical entity to a disease with heterogeneity in presentation, course and outcome. The phenotypic description in our study is similar to the earlier reported literature.
Cranial nerve involvement in GBS is common and a well-known fact, however there are no studies only focused on cranial nerve in GBS. Many of the clinicians don’t look for the cranial nerve involvement, until unless patient will complain about that, and hence less number of cases are picked up. If we are vigilant and look for cranial nerve pathology, the yield will be more and better care can be provided accordingly. In our study cranial nerve involvement was seen in 38 (62.3%) patients. Many of the studies done before quote variable involvement of cranial nerves ranging from 50% to 75%, for example, Lφeffel, et al.[9] have quoted 50% and Dhadke, et al.[10] had 62.5% involvement, which correlates well with our study. Majority of our patients (65%) had more than one cranial nerve palsies, suggesting a systemic insult. All of the patients with cranial nerve palsies had marked quadriparesis making them bed bound, suggesting severity of the illness was more in them. In our series few of them didn’t complain about symptomatology of cranial neuropathy, like in facial palsy out of 28 patients, 10 didn’t complain. On examination they found be having bilateral facial palsy of mild to moderate severity. Hence, dedicated history and examination is must to detect these cases.
Bulbar palsy was the most common (49.2%) in our study, which correlates with many studies. Among bulbar palsy patients majority had dysphagia and few had dysphonia. These patients were kept on Ryle's tube feeding, and in two patients it was an indication for intubation because of copious secretions. In the follow up at 1 month all of them had complete recovery.
Facial palsy was seen in 28 (46%) patients, all had bilateral involvement except 3, who had unilateral palsy. In many of the studies facial palsy was the most common finding with percentage ranging from 45% to 53%. In a study by Winer, et al.[11] 53% had bilateral facial palsy. In our patients three had unilateral palsy, which is rare but many case reports are there in the literature with unilateral palsy (Kamihiro, et al. and Verma, et al.).[1213] Only four patients complained about altered taste sensation, suggesting proximal involvement of facial nerve. At 1 month of follow up, 15 patients had partial recovery and rest had complete recovery. At the end of 3 months remaining 15 patients also had complete recovery.
Tongue paralysis was seen in 6 (10%) and ophthalmoplegia in 4 (6.5%) of our patients, which was similar to findings noted by Winer, et al.[11] In patients of ophthalmoplegia, their main complaint was diplopia in whom one had complete external ophthalmoplegia and remaining three had partial ophthalmoplegia. Deafness was seen in one patient, in whom audiogram revealed sensoryneural hearing loss in both ears. Although it's a rare finding, there are few case reports describing this entity (Takazawa, et al.)[14]. All these patients had multiple cranial nerve palsies including facial and bulbar palsies. Over 1 to 2 months these patients recovered completely.
On comparing various parameters among patients of GBS with and without cranial nerve involvement [Table 3], significant association was seen with respiratory paralysis, IVIg requirement and ventilator support among patients with cranial nerve palsies. Rest parameters didn’t show any correlation between the two groups, suggesting age, sex, antecedent illness and subtypes of GBS are not related to the presence or absence of cranial nerve palsies. Majority of bulbar palsy patients had respiratory paralysis, suggesting an indicator of respiratory involvement as mentioned in earlier studies. As compared to motor deficits, recovery of cranial nerve palsies is earlier.
We also found some rare cranial neuropathies, like involvement of vestibulocochlear in one patient, ophthalmoplegia in four and tongue paralysis in six patients. Through this again we want to emphasis that careful search should be done to detect these rare entities.
At the end of 6 month follow up, 75% were able to walk unassisted and only 25% were needing support to walk or dependent for their activities of daily living. A large prospective study by van Koningsveld, et al.[15] had almost similar results, 82% of their patients were able to walk independently at the end of 6 months. We didn’t find any difference in the recovery at the end of 6 months, in both the groups, which correlates well with the van Koningsveld, et al.[15] study.
Conclusion
Cranial neuropathy is common in GBS; careful search for this will yield more and rare cranial neuropathies. Our study found a correlation between cranial nerve palsies and severity of the illness. Bulbar palsy is an indicator of respiratory paralysis. Cranial nerve innervated muscles recover earlier as compared to distal limb muscles. No association was found between outcome at 6 months and cranial nerve involvement.
Acknowledgements
Dr. Bharat Bhushan, DM neuroresident, Dept of Neurology, Dr. S N Medical college, Jodhpur. Dr. Gaurav Kasundra, DM neuroresident, Dept of Neurology, Dr. S N Medical college, Jodhpur.
Source of Support: Nil.
Conflict of Interest: None declared.
References
- Differences in patterns of progression in demyelinating and axonal Guillain-Barré syndromes. Neurology. 2003;61:471-4.
- [Google Scholar]
- Clinical and epidemiological features of Guillain-Barré syndrome. J Infect Dis. 1997;176(Suppl 2):S92-8.
- [Google Scholar]
- A clinical and electrophysiologic survey of childhood Guillain-Barré syndrome. Pediatr Neurol. 2004;30:86-91.
- [Google Scholar]
- The Landry-Guillain-Barré syndrome; a clinicopathologic report of 50 fatal cases and a critique of the literature. Medicine (Baltimore). 1949;28:59-141.
- [Google Scholar]
- Guillain-Barre syndrome in northern China. Relationship to Campylobactor jejuni infection and anti-glycolipid antibodies. Brain. 1995;118:597-605.
- [Google Scholar]
- Sur un syndrome de radiculo-nevrite avec hyperalbuminose du liquid cephalorachidien sans reaction cellulaire. Remarques sur les characteres clinique et graphique des reflexes tendinaux. Bull Mem Soc Med Hop Paris. 1916;40:1462-70.
- [Google Scholar]
- The Landry-Gullian Barré syndrome. Complications, prognosis and natural history in 123 cases. J Neurol Sci. 1977;33:71-9.
- [Google Scholar]
- Clinical profile of Guillain Barre syndrome. J Assoc Physicians India. 2013;61:168-72.
- [Google Scholar]
- A prospective study of acute idiopathic neuropathy. I. Clinical features and their prognostic value. J Neurol Neurosurg Psychiatry. 1988;51:605-12.
- [Google Scholar]
- Acute motor-sensory axonal Guillain-Barré syndrome with unilateral facial nerve paralysis after rotavirus gastroenteritis in a 2-year-old boy. J Infect Chemother. 2012;18:119-23.
- [Google Scholar]
- Unilateral facial palsy in Guillain-Barre syndrome (GBS): A rare occurrence. BMJ Case Rep 2012 2012:bcr2012007077.
- [Google Scholar]
- Sudden deafness and facial diplegia in Guillain-Barré Syndrome: Radiological depiction of facial and acoustic nerve lesions. Intern Med. 2012;51:2433-7.
- [Google Scholar]
- A clinical prognostic scoring system for Guillain-Barré syndrome. Lancet Neurol. 2007;6:589-94.
- [Google Scholar]




