
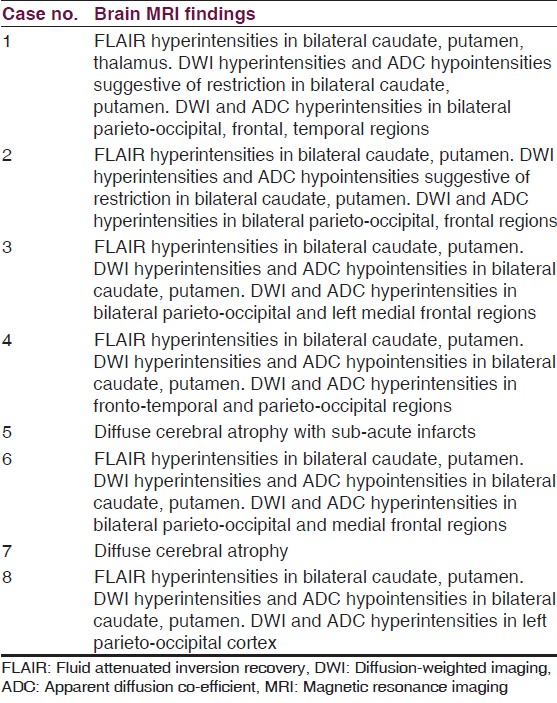
Translate this page into:
A study of clinical profile, radiological and electroencephalographic characteristics of suspected Creutzfeldt-Jakob disease in a tertiary care centre in South India
Address for correspondence: Dr. Rohan R. Mahale, Department of Neurology, MS Ramaiah Medical College and Hospital, Bangalore - 560 054, Karnataka, India. E-mail: rohanmahale83@gmail.com
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Introduction:
Creutzfeldt-Jakob disease (CJD) is a progressive, fatal, neurodegenerative disease classified under transmissible spongiform encephalopathies (TSE) or prion diseases. It is characterized by long asymptomatic period followed by rapid clinical deterioration leading to the death within months. The disease is still under-reported in India.
Objective:
The aim of this study was to describe the clinical, radiological and electroencephalographic characteristics of eight cases of CJD encountered in MS Ramaiah Medical college and Hospital, Bangalore over the past 3 years (2010-2013). This was retrospective, observational, hospital-based study.
Results:
The mean age of patients was 66.6 years (range: 54-82) and there was female predominance (five patients). The main clinical manifestations were cognitive disturbance (8/8) and myoclonus (8/8), followed by behavioral disturbance (5/8), ataxia (5/8) and extra-pyramidal symptoms/signs (4/8). Time interval (mean) between onset of disease to death was 6.6 months (range: 3-14). Brain MRI abnormalities were noted in 6 patients: Fluid-attenuated inversion recovery hyperintensities with restriction on diffusion-weighted image/apparent diffusion coefficient (DWI/ADC) in caudate and putamen, and diffusion hyperintensities without restriction on ADC in parieto-occipital, frontal and temporal regions. Classical electroencephalogram (EEG) changes of periodic triphasic waves were seen in 87% of patients. The CSF 14-3-3 protein assay was positive in two patients (out of four). Seven cases were probable CJD and one was possible CJD.
Conclusion:
A strong clinical suspicion aided by characteristic brain MRI and EEG abnormalities is essential for timely diagnosis of this fatal disease.
Keywords
Clinical manifestation
Creutzfeldt-Jakob disease
diagnosis
14-3-3 protein
prion diseases
prion protein

Introduction
Creutzfeldt-Jakob disease (CJD) is one of the transmissible spongiform encephalopathies (TSE) or prion diseases affecting human beings.[1] The other diseases are kuru, Gerstmann-Sträussler-Scheinker syndrome and fatal familial insomnia.[2] It is a fatal, neurodegenerative brain disease caused by abnormal accumulation and/or metabolism of a misfolded variant of the host-encoded prion protein (PrPC). The abnormally folded, insoluble isoform of PrPC, designated as prion scrapie protein (PrPSc), forms toxic aggregates in neurons causing spongiform (vacuolar) degeneration in the brain.[1] Clinically, the disease is characterized by a long asymptomatic period followed by a rapidly progressive clinical deterioration leading to death within months to few years. Both genders are equally affected with greatest frequency occurring in the age group of 45-75 years.[3] The main clinical symptoms consist of rapidly progressive dementia, myoclonus and cerebellar ataxia. Other symptoms include mood changes, psycho-affective and sleep disorders, tremors, visual hallucinations, cortical blindness and amyotrophy.[45] Sporadic CJD is the most common form of prion diseases accounting for 90% of cases.[6] The global annual incidence of CJD ranges from 0.3 to 1.1 per million populations.[7] In India, an incidence of 0.085 per million (8.5 cases in more than one billion population) is recorded by the CJD Registry at Department of Neuropathology, National Institute of Mental Health and Neurosciences, Bangalore (unpublished data). The tools for the clinical diagnosis of CJD include demonstration of altered signals on MRI diffusion-weighted imaging,[8] periodic sharp wave complexes (PSWC) on electroencephalography (EEG) recording and presence of the 14-3-3 protein in CSF.[9] In India, there are only two previous reports of CJD cases that include National CJD Registry at Department of Neuropathology, National Institute of Mental Health and Neurosciences, Bangalore,[10] and CJD case series from North India by Mehndiratta et al.[11] There is under-registration and recognition of the disease due to lack of strong clinical suspicion and paucity of centers or laboratories performing genetic and microbiological diagnostic tests. The study was aimed at providing more data on clinical, radiological and electroencephalographic characteristics of cases of CJD in India by reviewing cases encountered in our hospital over the past 3 years (2010-2013).
Materials and Methods
We performed an observational, descriptive, hospital-based study at the MS Ramaiah Institute of Neurosciences, MS Ramaiah Medical College and Hospital, Bangalore, India. The study was approved by the Institutional review board and ethical clearance was taken. Informed consent was taken at admission. All patients with rapidly progressive dementia admitted in the institute from June 2010 to June 2013 and diagnosed with CJD as per World Health Organization (WHO) diagnostic criteria for CJD[12] were included in the study. The disease was further classified into definite, probable or possible CJD [Table 1].

The files of included patients were reviewed in detail. The following data were recorded: Age at presentation, gender, occupation, dietary habits, duration of symptoms at time of diagnosis, clinical presentation at onset, subsequent symptoms/sign, history of surgical procedure or head trauma, past medical history, medications. Results of biological workup were also recorded: Full blood count, erythrocyte sedimentation rate (ESR), urinalysis, renal function tests including electrolytes and calcium, liver function test including plasma ammonia, anti-nuclear antibody (ANA), thyroid function tests including anti-thyroid peroxidase antibodies (anti-TPO) level, vitamin B12 , human immunodeficiency virus (HIV) screening test, serum Venereal Disease Research Laboratory (VDRL) test, voltage-gated potassium channel (VGKC) antibodies level, cerebrospinal fluid (CSF) analysis including cell count, cell type, protein, glucose, bacteriological and virological studies including 14-3-3 protein level in CSF. All patients underwent 1.5 Tesla brain magnetic resonance imaging (MRI) including diffusion-weighted (DWI) and fluid-attenuated inversion recovery (FLAIR) sequences. EEG was carried out using the international 10-20 system and EEG abnormalities was recorded in detail. The periodic sharp wave complexes (PSWC) were defined as complexes of biphasic or triphasic sharp waves with a duration of 100-600 ms, recurring every 500 to 2000 ms.[2] The final clinical outcome of each patient was recorded.
Results
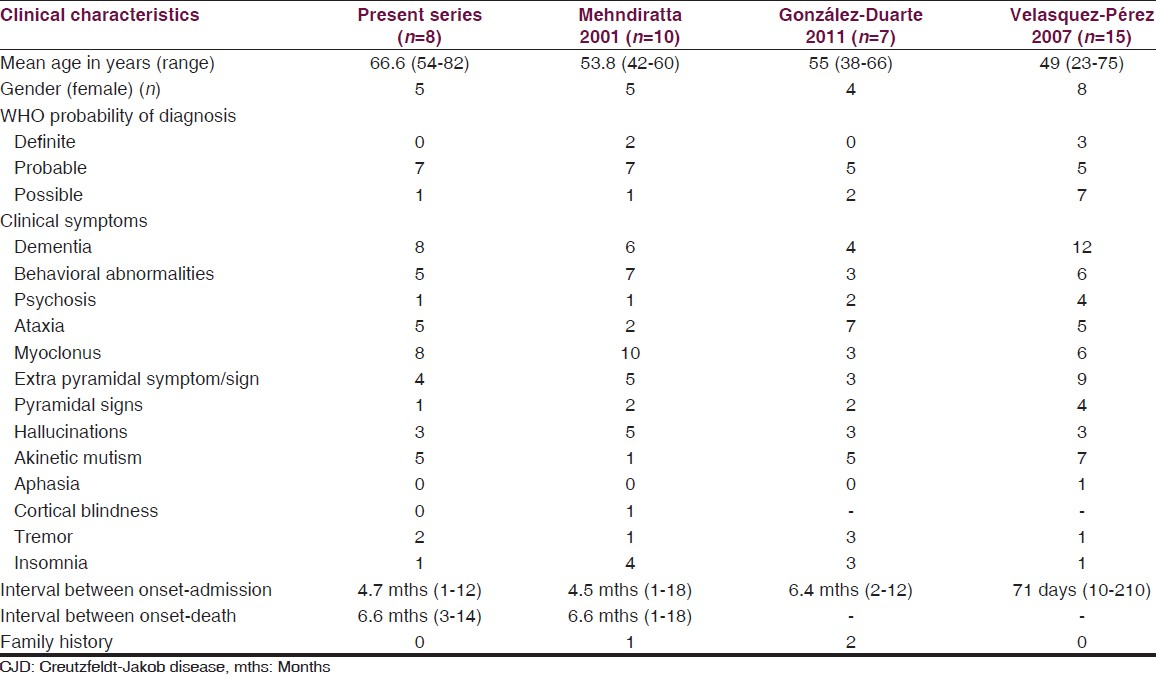
By screening the hospital records for the diagnosis of CJD during the study period, we found eight cases satisfying the WHO diagnostic criteria for CJD. The mean age of patients at presentation was 66.6 years (range: 54-82). Five patients were females (F:M = 5:3) and were housewives except one who was a nurse. Seven of these cases were probable CJD and one was possible CJD. Demographic, clinical and paraclinical charac-teristics of cases are summarized in Table 2. The detailed clinical history, laboratory investigation, radiological and electroencephalographic data of each patient is illustrated in supplementary material. Two patients had history of surgical procedures in the past (coronary artery bypass graft and gastric antrectomy respectively). None of the patient had head injury in the past. The main clinical manifestations were cognitive disturbance (8/8) and myoclonus (8/8), followed by behavioral disturbance (5/8), ataxia (5/8) and extra pyramidal symptoms/signs (4/8).

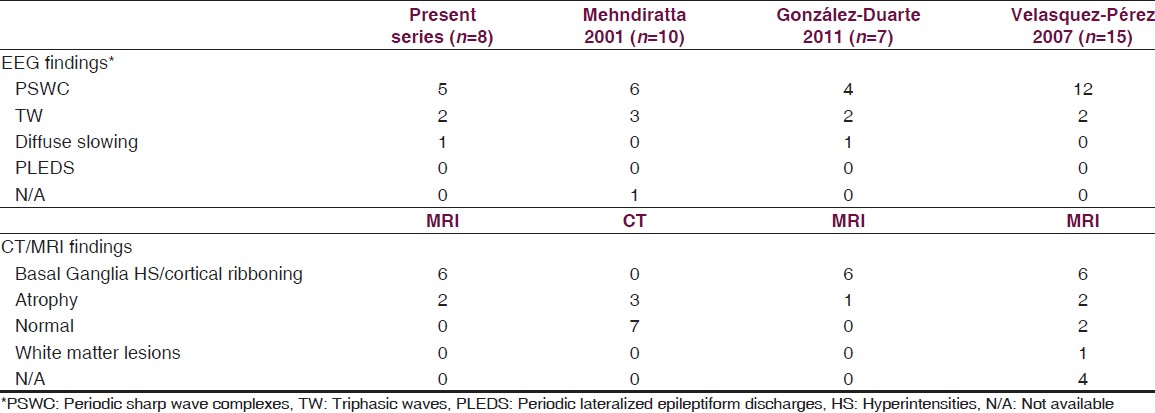
The first symptoms at onset were memory impairment/dementia (3/8), ataxia (1/8), behavioral disturbances (1/8), psychosis (1/8), extrapyramidal symptoms (1/8) and pyramidal signs (1/8). In one patient, the disease started as psychosis with history of delusions and hallucinations and was treated with anti-psychotics before the full picture of CJD evolved. Five patients rapidly progressed to a state of akinetic mutism (5/8). The mean time interval between symptoms onset to admission was 4.7 months (range: 1-12) and mean time interval between symptoms onset to death was 6.6 months (range: 3-14). None of the patients had positive family history of similar disease. Brain MRI abnormalities were noted in six of our eight patients of suspected CJD. In six patients, brain MRI FLAIR showed hyperintensities in caudate on both sides, putamen. One patient had FLAIR hyperintensity in thalamus with no diffusion restriction. DWI hyperintensities and apparent diffusion coefficient (ADC) hypointensities were seen in caudate, putamen in all six patients. DWI hyperintensities and ADC hyperintensities were noted in parieto-occipital (six patients), frontal (five patients) and temporal regions (two patients). There was reduced diffusion in the basal ganglia only but not in the cortices. In two patients, brain MRI showed only cortical atrophy. Figures 1 and 2 show the characteristic brain MRI abnormalities of selected cases. Brain MRI characteristics of each patient are summarized in Table 3. CSF analysis was carried out in all patients, which showed normal to mildly elevated protein, cell count within normal limits and negative bacteriological/virological studies. The CSF 14-3-3 protein assay was carried out in four patients and was positive in two patients of them. Serum VGKC antibodies were assessed in five patients and were negative. Classical EEG abnormalities in the form of PSWC's were seen in five patients (5/8). EEG showed triphasic waves in two patients and diffuse slowing of background rhythm in one patient. Figure 3 shows the characteristic EEG abnormalities of selected cases. Two patients were given methylprednisolone with clinical suspicion of immune mediated encephalopathy but with no improvement. Postmortem brain examination for confirmation of diagnosis could not be carried out in any of the patients. A comparison of clinical, radiological and electroencephalographic characteristics with other CJD case series is illustrated in Tables 4 and 5.

- (a) Axial FLAIR MRI brain (patient 2) showing both caudate and putamen hyperintensities (arrow), (b) DWI (patient 2) showing hyperintensity in both basal ganglia (arrow), frontal, and parieto-occipital regions, (c) ADC (patient 2) showing hypointensity in both basal ganglia (arrow), (d) DWI (patient 3) showing hyperintensity in both caudate (arrow), frontal, and parieto-occipital regions, (e) ADC (patient 3) showing hypointensity in bilateral caudate (arrow), (f) DWI (patient 3) showing hyperintensity in frontal and parieto-occipital regions (arrow), (g) ADC (patient 3) showing hyperintensity in frontal and parieto-occipital regions (arrow)

- (a) Axial FLAIR MRI brain (patient 4) showing bilateral caudate, putamen hyperintensities (arrow) and fronto-temporal region, (b) DWI (patient 4) showing hyperintensity in bilateral caudate, putamen (arrow), fronto-temporal and parieto-occipital region, (c) ADC (patient 4) showing hypointensity in bilateral caudate (arrow), (d) DWI (patient 8) showing hyperintensity in bilateral caudate (arrow), left parieto-occipital region, (e) ADC (patient 8) showing hypointensity in left caudate (arrow)
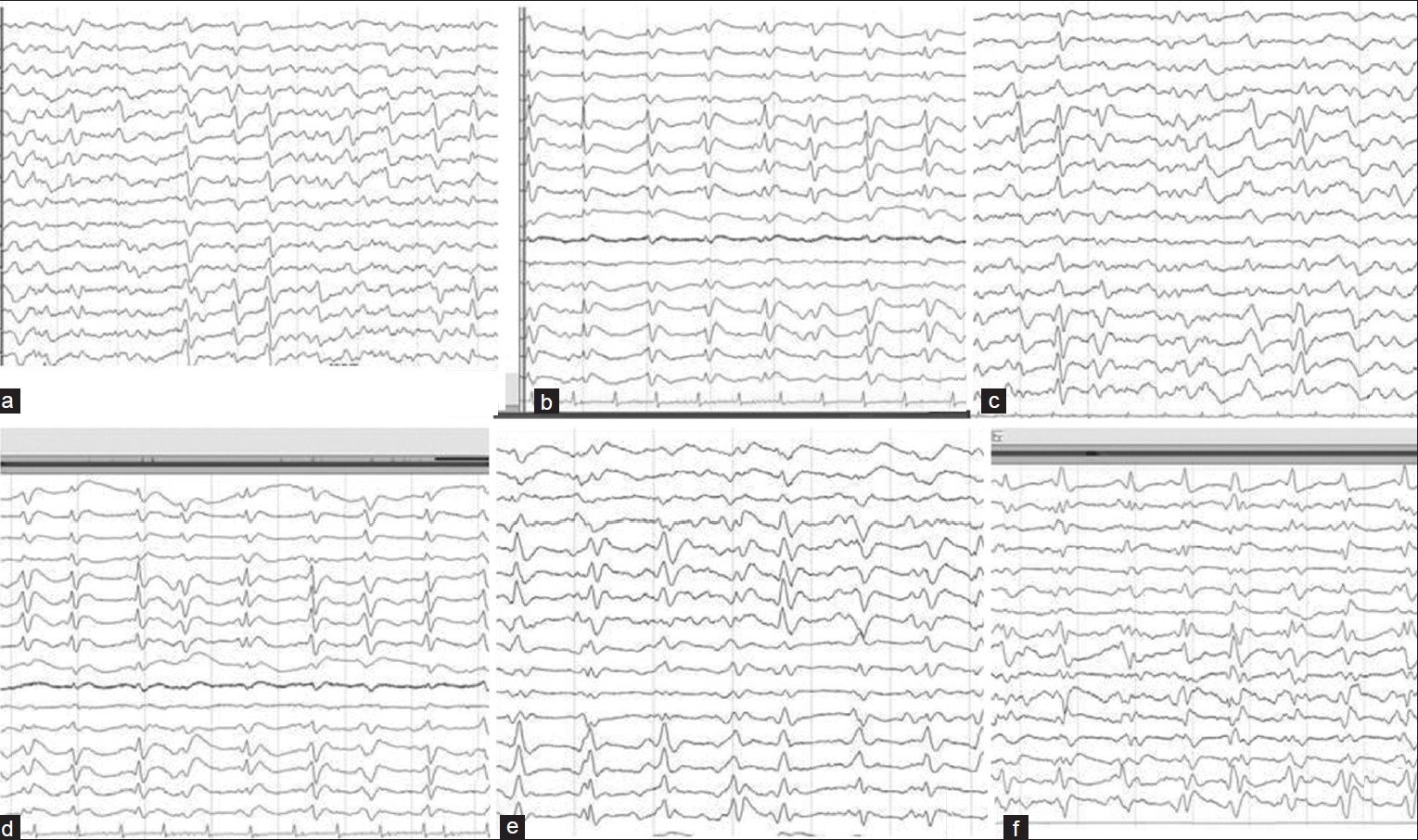
- (a, c-e) EEG showing periodic sharp wave complexes at 1-2/sec with slowing of background rhythm. (b and f) EEG showing bilateral triphasic waves (TW) predominant in frontal channels with background slowing
Discussion
CJD is a human prion disease that has a long asymptomatic period and a fatal outcome. Published cases of CJD in India include 85 cases from the National CJD registry, NIMHANS, Bangalore (1968-2005)[10] and 10 cases from North India reported by Mehndiratta et al.[11] (1990-1998). Eight cases were diagnosed at our institution in the previous 3 years. The mean age of the patients was 66.6 years, which is similar to the published mean age for sporadic CJD in the literature.[35] In the case series by Mehndiratta et al.,[11] patients seemed to be younger (53.8 years). Similarly, the mean age of patients was younger in other case series.[513] We need to be cautious when comparing mean age between studies as our sample size is small and the apparent differences in the mean age are due to random fluctuations. We found female predominance (5/8) in our series as in other published case series.[51114] All of our patients had rapidly progressive dementia with myoclonus. Other clinical manifestations were behavioral disturbances, ataxia, and extrapyramidal symptoms. Similar observations were noted in the case series by Velasquez-Perez et al.[5] and González-Duarte et al.[14] All of our patients were evaluated with other relevant investigations to rule out other causes for encephalopathy like infective, metabolic, paraneoplastic and autoimmune etiologies.
Behavioral disturbances in CJD occur in 30% of patients at the onset of disease and in 57% at later stages.[6] It was seen in 62% of patients in our series. None of the patients had a family history of similar disease. Thus, it is very likely that our eight patients are sporadic CJD. González-Duarte et al.[14] had reported two patients and Mehndiratta et al.[11] reported one patient having family history. The time interval between the onset of symptoms and diagnosis varied from 1 month to 12 months with mean of 4.7 months which was similar to findings in other case series.[51114] The mean duration from symptom onset to death was 6.6 months (range: 3-14), similar to other case series.[51114] In the literature, the mean survival of CJD patients is 5 months and about 80% of patients succumb to disease after 12 months from onset.[6]
The brain MRI changes in CJD has been demonstrated to precede EEG or CSF abnormalities.[15] However, it may be without abnormality early in the disease course.
High signals in T2-weighted/FLAIR sequences have been linked to astrogliosis.[16] Isolated cortical hyperintensity and combined cortical and deep gray matter (basal ganglia) hyperintensity are the two patterns of DWI and/or FLAIR abnormality that have been described.[17] DWI is more sensitive than FLAIR in the detection of cortical abnormalities in early stages of CJD. Vacuole formation is the principal neuropathologic finding in CJD and they restrict diffusion of water resulting in DWI hyperintensity. DWI has higher sensitivity (92%) and specificity (93%) in the diagnosis of CJD regardless of PSWCs.[18] Involvement of deep gray matter is associated with shorter disease course with rapidly progressing neurologic deterioration whereas absence of basal ganglia involvement correlates with delayed onset of dementia and longer disease course.[19]
Our six patients had hyperintensities in caudate and putamen on FLAIR images which were hyperintense on DWI images and hypointense on ADC images suggesting diffusion restriction. DWI hyperintensities were noted in parieto-occipital in all six, frontal in five and temporal region in two patients. But the corresponding ADC maps in these cortical regions were hyperintense suggesting no diffusion restriction. Our patients had only basal ganglia diffusion restriction. Similarly, brain MRI of seven patients of CJD studied by González-Duarte et al. (2011) had abnormalities in five patients. All five patients had bilateral basal ganglia hyperintensities on FLAIR with diffusion restriction with no cortical hyperintensities or restriction.[14] However, brain MRI of 10 patients of CJD studied by Biswas et al.(2013) had abnormalities in all 10 patients. Eight patients had bilateral basal ganglia FLAIR hyperintensities with diffusion restricition. Six patients had diffusion restriction in parieto-occipital and two patients in frontal region.[20]
A DWI and FLAIR combination has higher sensitivity (91%) and specificity (95%) for CJD.[21] However, Vitali et al. (2011) drew a conclusion that the pattern of FLAIR/DWI hyperintensity and reduction of ADC in striatal hyperintensity regions on DWI differentiates sporadic CJD from other rapidly progressive dementia with 98% sensitivity (95% CI 0.89-1.00) and 100% specificity (95% CI 0.88-1.00).[22]
Brain MRI of two patients showed only cortical atrophy without classical changes of CJD. One patient was diagnosed to have extrapyramidal syndrome with dementia (Case 5) and other patient was diagnosed as paranoid schizophrenia with recurrent hyponatraemia (Case 7). The reason for MRI negativity were: Firstly, the duration of illness in these two patients were longer (12 months) and secondly, the brain MRI was done during the terminal stage of illness (10-11 months after onset) wherein the typical MRI abnormalities might have disappeared.[2324]
The classical EEG changes (PSWC and triphasic waves) were observed in all the patients except in one, whose EEG showed diffuse slowing of background activity. EEG has a sensitivity of 67% and specificity of 74-86% in the diagnosis of CJD.[1423] Repeated EEG during the course of disease increases the probability of demonstrating characteristic EEG abnormality.[25] EEG abnormalities are rarely seen in patients with other cause of dementia like Alzheimer's or vascular dementia.
CSF examination was carried out in all patients and did not reveal pleocytosis. The detection of the 14-3-3 protein in CSF has been one of the markers for diagnosis of CJD.[26] They are probably moderately accurate in diagnosing sporadic CJD with sensitivity of 92% (95% confidence interval [CI] 89.8-93.6) and specificity 80% (95% CI 77.4-83.0).[27] But Zerr et al. (2009) refuted this observation as they found that MRI findings are equivalent to elevated levels of the 14-3-3 protein in the diagnosis of probable sporadic CJD.[28] Other biomarkers such as t-tau, p-tau, S-100, or neuron-specific enolase (NSE) in the CSF are required in addition to, or in lieu of, protein 14-3-3.[27] In our series, the assay was carried out only in four cases and was positive in two cases.
This case series represents a single-center experience from south India. CJD is still considered a rare disease in India. Apart from National CJD registry, NIMHANS and case series from New Delhi, there are only few case reports of CJD. All our eight patients were resident of Bangalore city, Karnataka. This geographical clustering assumes significance. This study demonstrates the fact that CJD is prevalent in India. There is need for spreading awareness in India about early suspicion and recognition of this fatal disease among the treating general medical practitioner, physicians and psychiatrist. The prompt referral to tertiary center with neuroscience expertise is also warranted. The CJD surveillance in India has to be improved by means of availability of sophisticated tests like CSF biomarkers, facilities for antemortem diagnosis of CJD and prion protein genotyping which is lacking.
The present case series had its own limitation. First, postmortem brain examination for confirmation of diagnosis could not be carried out in any of the patients. Histological examination and immunostaining for protease-resistant protein (PrPSc ) are the gold standard for the diagnosis. As a result, all our cases were suggestive of prionopathy due to lack of histological confirmation. Second, PrP genotyping/polymorphism of codon 129 of the prion protein gene (PRNP) of the patients could not be carried out due to lack of laboratory facilities. This is important as differences in the polymorphic codon 129 and PrPSc typing is linked to clinical heterogeneity of sporadic CJD. Third, the 14-3-3 protein assay could not be carried out in all patients due to financial constraints, lack of local expertise and logistic problems for transferring to a foreign laboratory.
Conclusions
CJD is fatal neurodegenerative disease. It has a wide range of clinical manifestations including cognitive decline, myoclonus, ataxia, pyramidal and extrapyramidal signs/symptoms, behavioral abnormalities and psychosis. Patient presenting with rapidly progressive dementia with myoclonus points toward CJD as one of the diagnosis. MRI brain in particular DWI and ADC abnormalities help in the early diagnosis. EEG abnormalities as well as CSF biomarkers like 14-3-3 protein, CSF-tau and neuron-specific enolase level help for the diagnosis. A strong clinical suspicion together with characteristic MRI and EEG abnormalities despite low availability of the CSF 14-3-3 protein assay are essential in timely diagnosis of this fatal disease.
Source of Support: Nil.
Conflict of Interest: None declared.
References
- Human prion diseases: Epidemiology and integrated risk assessment. Lancet Neurol. 2003;2:757-63.
- [Google Scholar]
- Manual for Surveillance of Human Transmissible Spongiform Encephalopathies Including Variant Creutzfeldt-Jakob Disease. Geneva: World Health Organization; 2003.
- Creutzfeldt-Jakob disease and related transmissible spongiform encephalopathies. N Engl J Med. 1998;339:1994-2004.
- [Google Scholar]
- Human spongiform encephalopathy: The National Institutes of Health series of 300 cases of experimentally transmitted disease. Ann Neurol. 1994;35:513-29.
- [Google Scholar]
- Creutzfeldt-Jakob disease: Patterns of worldwide occurrence and the significance of familial and sporadic clustering. Ann Neurol. 1979;5:177-88.
- [Google Scholar]
- Accuracy of diffusion-weighted MR imaging in the diagnosis of sporadic Creutzfeldt-Jakob disease. J Neurol. 2003;250:222-5.
- [Google Scholar]
- Challenging the clinical utility of the 14-3-3 protein for the diagnosis of sporadic Creutzfeldt-Jakob disease. Arch Neurol. 2003;60:813-6.
- [Google Scholar]
- Creutzfeldt-Jakob disease: Report of 10 cases from North India. Neurol India. 2001;49:338-41.
- [Google Scholar]
- WHO Consultation on Global Surveillance, Diagnosis and Therapy of Human Transmissible Spongiform Encephalopathies. In: WHO/EMC/ZDI/98.9. Geneva: WHO; 1998.
- [Google Scholar]
- Accuracy and reliability of periodic sharp wave complexes in Creutzfeldt-Jakob disease. Arch Neurol. 1996;53:162-6.
- [Google Scholar]
- Can prion disease suspicion be supported earlier. Clinical, radiological and laboratory findings in a series of cases? Prion. 2011;5:201-7.
- [Google Scholar]
- Correlation of diffusion-weighted magnetic resonance imaging with neuropathology in Creutzfeldt-Jakob disease. Arch Neurol. 2002;59:128-34.
- [Google Scholar]
- MRI in sporadic Creutzfeldt-Jakob disease: Correlation with clinical and neuropathological data. Neuroradiology. 1998;40:65-70.
- [Google Scholar]
- Isolated cortical signal increase on MR imaging as a frequent lesion pattern in sporadic Creutzfeldt-Jakob disease. AJNR Am J Neuroradiol. 2008;29:1519-24.
- [Google Scholar]
- Diffusion-weighted MRI abnormalities as an early diagnostic marker for Creutzfeldt-Jakob disease. Neurology. 2004;10:443-9.
- [Google Scholar]
- Sporadic Creutzfeldt-Jakob disease: Magnetic resonance imaging and clinical findings. Neurology. 2004;63:450-56.
- [Google Scholar]
- Case series of probable sporadic Creutzfeldt-Jakob disease from Eastern India. Ann Indian Acad Neurol. 2013;16:659-63.
- [Google Scholar]
- Diffusion-weighted and fluid-attenuated inversion recovery imaging in Creutzfeldt-Jakob disease: High sensitivity and specificity for diagnosis. AJNR Am J Neuroradiol. 2005;26:1551-62.
- [Google Scholar]
- Diffusion-weighted MRI hyperintensity patterns differentiate CJD from other rapid dementias. Neurology. 2011;76:1711-9.
- [Google Scholar]
- Diffusion-weighted imaging and magnetic resonance spectroscopy of sporadic Creutzfeldt-Jakob disease: Correlation with clinical course. Neuroradiology. 2011;53:939-45.
- [Google Scholar]
- Creutzfeldt-Jakob disease: Serial changes on diffusion-weighted MRI. J Comput Assist Tomogr. 2001;25:274-7.
- [Google Scholar]
- Serial EEG findings in 27 cases of Creutzfeldt-Jakob disease. Arch Neurol. 1980;37:143-5.
- [Google Scholar]
- Analysis of EEG and CSF 14-3-3 proteins as aids to the diagnosis of Creutzfeldt-Jakob disease. Neurology. 2000;55:811-5.
- [Google Scholar]
- Evidence-based guideline: Diagnostic accuracy of CSF 14-3-3 protein in sporadic Creutzfeldt-Jakob disease: Report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2012;79:1499-506.
- [Google Scholar]
- Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Brain. 2009;132:2659-68. F
- [Google Scholar]
Supplement
Following are the illustration of each case in detail.
Case 1
A 64-year-old right-handed housewife who is a vegetarian was brought to our hospital with history of memory disturbance of 2 months duration with rapid worsening in her mental status of 2 weeks duration.
Memory disturbance was mainly for recent events. Along with memory disturbance, the patient also had history of gait unsteadiness of 1 month duration. The patient deteriorated rapidly 2 weeks prior to her hospital admission. The patient became bed bound with refusal of food, difficulty in swallowing, doubly incontinent. The patient relatives also noted jerky movement of body and limbs to acoustic stimulus suggestive of stimulus sensitive myoclonus. There was no preceding history of fever, headache, vomiting or seizures. No dog bite, head injury or recent immunization. She was diabetic and hypertensive. She was diagnosed with gastric antral carcinoma 3 years back and had undergone gastrectomy with one cycle of chemotherapy. At admission, she was drowsy but arousable, not obeying verbal commands, used to have startle myoclonus on arousal. There was no motor weakness but had rigidity of all four limbs with preserved deep tendon reflexes. Gait could not be assessed. Her complete hemogram, renal function test, liver function test including ammonia, thyroid function test, serum electrolytes including calcium were within normal limits. Serum anti-TPO antibodies were 26 IU/L. The patient had a low vitamin B12 and vitamin D3. ANA, VGKC and NMDA receptor antibody were negative. Her serum ACE level was within normal limits. She was seronegative for HIV and serum VDRL. CSF analysis showed protein of 36 mg/dL, glucose of 81 mg/dL with 2 cells/cumm (lymphocytes). CSF gram stain, AFB stain, cryptococcal antigen, TB-PCR and HSV-DNA PCR was negative. CSF 14-3-3 protein was less than 1.0 ng/ml (reference range <1.5 ng/ml). CSF paraneoplastic panel was negative for following antibodies-antineuronal nuclear antibodies (ANNA-1, 2, 3), Purkinje cell cytoplasmic antibodies (PCA.1, 2), anti-amphiphysin, CRMP-5, anti Ma/Ta and anti-yo antibodies. The brain MRI showed T2/FLAIR hyperintensities in bilateral caudate, putamen, thalamus with diffusion hyperintensities in bilateral parieto-occipital, frontal, temporal region and restriction in bilateral caudate, putamen. EEG showed generalized, periodic sharp waves of diphasic and triphasic morphology occurring at 1 Hz without anterior to posterior lag. The patient was diagnosed as probable CJD in view of clinical symptoms of rapidly progressive dementia, myoclonus with ataxia, duration of disease less than 2 years and EEG abnormality. MRI brain abnormality was compatible with diagnosis of CJD. However, CSF 14-3-3 protein level was within normal range. The patient died within 2 months after being discharged.
Case 2
An 82-year-old right-handed man presented with history of gait unsteadiness of 5 months duration, blurring of vision with recent memory loss, visuo-spatial disorientation in the form of getting confused between rooms of his house, dressing apraxia and behavioral disturbance in the form of emotional lability of 4 months duration. Subsequently, he developed multifocal myoclonic jerks of extremities which were stimulus sensitive since 1 month before presenting to our hospital along with slurring of speech of 1 week duration. There was history of weight loss and insomnia. The patient was also incontinent. The patient used to consume vegetarian diet. No other co-morbidities. On examination, the patient was conscious, oriented to time but not to place. His MMSE score was 23/30. Neuropsychological assessment revealed bilateral frontal and right parietal lobe involvement. He had an ataxic dysarthria. No cranial nerve involvement or eye movement abnormalities. Muscle strength was normal and muscle stretch reflexes were sluggish. Plantar response was flexor. There was stimulus sensitive myoclonus to sudden acoustic stimulus. There was evidence of appendicular ataxia (left > right) with gait ataxia. Routine hematological and biochemical parameters including ammonia and thyroid functions were within normal limits. Serum anti-TPO antibodies were 38 IU/L. The patient had a low vitamin B12 and vitamin D3. ANA and VGKC antibody were negative. He was seronegative for HIV and serum VDRL. CSF analysis showed protein of 46 mg/dL and glucose of 66 mg/dL with 2 cells/cumm (lymphocytes). CSF gram stain, cryptococcal antigen, TB-PCR and HSV-DNA PCR was negative. CSF 14-3-3 protein was high >28 ng/ml. The brain MRI showed T2/FLAIR hyperintensities in bilateral caudate, putamen, with diffusion hyperintensities in bilateral parieto-occipital, frontal regions and restriction in bilateral caudate. EEG showed bifrontal predominant periodic sharp waves occurring at 1 Hz. The patient was diagnosed as probable CJD in view of clinical symptoms of rapidly progressive dementia, myoclonus with ataxia, duration of disease less than 2 years and EEG abnormality. MRI brain abnormality was compatible with diagnosis of CJD. The patient died within 1 month after being discharged.
Case 3
A 73-year-old housewife was brought by her family members with the history of disturbance in recent memory-used to forget recent events, delayed response time and difficulty in recalling familiar faces and names of 2 months duration. There was history of behavioral disturbance in the form of agitated behavior, irritability with emotional lability of 15 days duration. She also had gait unsteadiness of 10 days duration. There was infrequent brief, jerky movement of limbs. No febrile illness, recent vaccination, head injury or surgical procedures. She was diabetic, hypertensive and had ischemic heart disease. At admission, she was afebrile, drowsy but arousable and was obeying simple verbal commands. She had ataxic dysarthria. Motor examination was normal with plantars flexor. There was stimulus sensitive myoclonus with severe ataxic gait. Routine hematological and biochemical parameters including ammonia, vitamin B12 and thyroid functions were normal. Serum anti-TPO antibodies and ACE level were normal. ANA and VGKC receptor antibody were negative. She was seronegative for HIV and VDRL. CSF analysis showed protein of 42 mg/dL and glucose of 72 mg/dL with 1 cell/cumm (lymphocytes). CSF gram stain, cryptococcal antigen, TB-PCR and HSV-DNA PCR were negative. CSF 14-3-3 protein was less than 1.0 ng/ml (reference range <1.5 ng/ml). The brain MRI showed T2/FLAIR hyper intensities in bilateral caudate, putamen, with diffusion restriction in the above-mentioned areas, diffusion hyperintensities in bilateral parieto-occipital and left medial frontal region. EEG was abnormal, showing diffuse slowing of background rhythm (4.5 Hz) with periodic sharp wave discharge from the left cerebral hemisphere. The patient was diagnosed as probable CJD in view of clinical symptoms of rapidly progressive dementia, myoclonus with ataxia, akinetic mutism, and duration of disease less than 2 years with EEG abnormality. MRI abnormality was compatible with the diagnosis of CJD. The patient died within 2 months after being discharged.
Case 4
A 64-year-old right-handed man developed clumsiness with incoordination in the right hand of 8 weeks duration. He had difficulty performing tasks like eating with spoon, opening bottles and buttoning/unbuttoning his shirt. Two weeks later, patient noted brief, jerky movement of right upper limb, initially during action, later during rest and to startle. Two weeks after right upper limb involvement, he developed similar jerky movement of left upper limb, both during rest and action. Patient family members also noted that he was forgetting recent events occurring in his house. One week later, the patient noted difficulty in walking with sense of imbalance requiring wall support to walk with tendency to fall sideways and slurring of speech. He did not have acral paraesthesia in limbs, tinnitus, visual disturbances or hard of hearing. No preceding febrile illness, drug intake, toxin exposure, head injury or surgical procedures. The patient was a hypertensive with no other co-morbidities. At admission, the patient was conscious, oriented. His attention span was reduced. MMSE was 23/30. Neuropsychological assessment revealed involvement of left dorsolateral prefrontal cortex. There was no eye movement abnormality. His speech was slurred suggestive of ataxic dysarthria. There was startle myoclonus involving limbs. Motor examination revealed asymmetric rigidity of right upper and lower limb with brisker deep tendon reflexes. However, plantars were flexor. There was appendicular ataxia (right > left) with severe gait ataxia. Routine hematological and biochemical parameters including ammonia, vitamin B12 and thyroid functions were normal. Serum anti-TPO antibodies and ACE level were normal. ANA and VGKC receptor antibody were negative. He was seronegative for HIV and VDRL. CSF analysis showed protein of 55 mg/dL and glucose of 76 mg/dL with 1 cell/cumm (lymphocytes). CSF gram stain, cryptococcal antigen, TB-PCR and HSV-DNA PCR were negative. EEG was abnormal, showing diffuse, asymmetrical slowing of background rhythm left > right (4.5 Hz). The brain MRI showed cerebral atrophy. The patient was treated with five doses of intravenous methylprednisolone but had no improvement in the symptomatology. He was discharged at request. At 1 month follow up, he had deteriorated and became bed bound, akinetic mute with incontinence. Repeat MRI brain showed T2/FLAIR hyperintensities in bilateral caudate, putamen with diffusion restriction in the same areas and diffusion hyperintensities in fronto-temporal and parieto-occipital regions. The patient was diagnosed as possible CJD in view of clinical symptoms of rapidly progressive ataxia, myoclonus with dementia, duration of disease less than 2 years and MRI abnormality. He succumbed to death within 1 month of follow up.
Case 5
A 67-year-old housewife was brought to the hospital with symptoms of decreased responsiveness, drowsiness, and incontinence of 1 week duration. On reviewing her history, she had developed progressive slowness in walking and day to day activities over the last 1 year. Her family members also noted memory disturbances in the form of forgetfulness since 6 months. She had developed behavioral disturbances in the form of irritability, anger outburst, excessive crying since 3 months. 1 month prior to present admission, she was wheel chair bound, requiring assistance for her activities of daily living. She used to have brief, jerky extremities of limb which was stimulus sensitive and her speech was slurred. Since 1 week, she had developed drowsiness, incontinence. No preceding febrile illness, seizures or fall. She is diabetic as well as hypertensive. At admission, the patient was drowsy, arousable. She had ataxic dysarthria with eye movements normal. Motor system examination revealed rigidity in all the limbs with resting tremors. In addition, there was startle myoclonus on examination. Gait could not be assessed. Routine hematological and biochemical parameters including ammonia, vitamin B12 and thyroid functions were normal. Serum anti-TPO antibodies and ACE level were normal. ANA and VGKC receptor antibody were negative. She was seronegative for HIV and VDRL. CSF analysis showed protein of 48 mg/dL, and glucose of 83 mg/dL with 1 cell/cumm (lymphocytes). CSF gram stain, cryptococcal antigen, TB-PCR and HSV-DNA PCR were negative. EEG was abnormal, showing diffuse slowing of background rhythm (4.5 Hz) with generalized, periodic sharp wave discharge occurring at 1 per second. The brain MRI showed diffuse cerebral atrophy with sub-acute infarcts. During hospital stay, her sensorium worsened over days as she became stuporous with frequent, multi-focal myoclonus. She succumbed to death within 4 weeks of admission. The patient was diagnosed as probable CJD in view of clinical symptoms of rapidly progressive dementia, myoclonus with extrapyramidal symptoms/ signs, and duration of disease less than 2 years with EEG abnormality.
Case 6
A 62-year-old right-handed man was brought to the emergency department of our hospital with the complaints of behavioral change of 1 month duration. 8 months back, the patient had a cerebro-vascular accident. Left MCA territory infarct and had made a significant recovery, minimally dependent for activities of daily living. Until 2 months before presenting to hospital, the patient was carrying out his day to day activities very well. Since 2 months, according to the close family members, the patient started having memory disturbances in the form of forgetfulness, unable to remember recent events. He started making errors in his office work. He developed behavioral disturbance of 1 month duration. The patient used to get agitated for trivial issues, irritable, episodes of anger outburst, emotionally labile with history of visuo-perceptual disturbance in the form of illusions and visual hallucination. There was history of startle myoclonus of 2 weeks duration. Since 1 week, the patient is bed bound, refusing feeds and incontinent. No febrile illness, dog bite, immunization. The patient had undergone CABG few years back. He was diabetic and hypertensive. At admission, the patient was conscious, not obeying simple verbal commands, bed bound. No neck stiffness. No startle myoclonus. Motor system was normal with plantars flexor. Routine hematological and biochemical parameters including ammonia, vitamin B12 and thyroid functions were normal. Serum anti-TPO antibodies and ACE level were normal. ANA and VGKC receptor antibody were negative. He was seronegative for HIV and serum VDRL. CSF analysis showed protein of 46 mg/dL and glucose of 68 mg/dL with 1 cell/cumm (lymphocytes). CSF gram stain, cryptococcal antigen, TB-PCR and HSV-DNA PCR were negative. The brain MRI showed T2/FLAIR hyperintensities in bilateral caudate, putamen with diffusion restriction in the same areas, and diffusion hyperintensities in bilateral parieto-occipital and medial frontal regions. EEG was abnormal, showing generalized, periodic sharp wave discharge of diphasic and triphasic morphology occurring at 1 per second. The patient was diagnosed as probable CJD in view of clinical symptoms of rapidly progressive dementia, myoclonus with visuo-perceptual disturbance, duration of disease less than 2 years and EEG abnormality. MRI brain abnormality was compatible with CJD. The patient was discharged and at 1 month follow up, he was bed bound, akinetic mute with frequent multifocal myoclonus and incontinent. He succumbed to death within 2 months of follow up.
Case 7
A 67-year-old, right-handed nurse was brought with history of behavioral disturbance of 1 year duration. To start with, she used to have suspicion toward her family members with episodes of anger outburst. Later, she started having auditory hallucination including third person hallucination as well as visual hallucination. She used to have persecutory delusion. For the same, she was evaluated by psychiatrist and was on anti-psychotics for few months. Subsequently 6 months later, she developed episodes of encephalopathy. On evaluation for the same, was found to have hyponatremia. She used to recover on correction of hyponatremia. Since 3 months, patient family members noted that she had progressive cognitive decline with gait unsteadiness. She used to sway on both side and fall. She stopped cooking and was wheel chair bound. 1 month before admission, her condition deteriorated, she became bed bound, mute with fluctuation in sensorium and incontinence. She used to have brief, jerky movements of limbs. No febrile illness, head injury or surgical procedures. She was diabetic with no other co-morbidities. At admission, she was drowsy, but arousable, not obeying verbal commands, used to have startle myoclonus on arousal. No eye movement restriction. There was no motor weakness but had rigidity of all four limbs with preserved deep tendon reflexes. Gait could not be assessed. Her complete hemogram, renal function test, liver function test including ammonia, thyroid function test and serum calcium were within normal limits. Her serum sodium was low (126 mEq/L). Serum anti-TPO antibodies were 33 IU/L. The patient had a low vitamin B12 and vitamin D3. ANA, VGKC antibody was negative. Her serum ACE level was within normal limits. She was seronegative for HIV and serum VDRL. CSF analysis showed protein of 41 mg/dL, glucose of 76 mg/dL with 3 cells/cumm (lymphocytes). CSF gram stain, AFB stain, cryptococcal antigen, TB-PCR and HSV-DNA PCR were negative. CSF 14-3-3 protein level was high > 100 ng/ml (reference range < 1.5 ng/ml). EEG showed generalized, periodic sharp waves of diphasic and triphasic morphology occurring at 1 Hz without anterior to posterior lag. The brain MRI showed diffuse cerebral atrophy with prominent ventricles. Her serum sodium was corrected and EEG was repeated three times during her hospital stay, but the EEG abnormalities persisted. The patient was diagnosed as probable CJD in view of clinical symptoms of rapidly progressive dementia, myoclonus with ataxia, duration of disease less than 2 years and EEG abnormality. CSF 14-3-3 protein level was positive which was compatible with the diagnosis of CJD. The patient succumbed to death within 2 months of admission.
Case 8
A 54-year-old housewife was brought with history of behavioral change in the form of irritability, anger outburst and excessive crying, along with cognitive decline of 1 month duration. The patient was not able to recognize relatives, used to forget recent events and was unable to carry her activities of daily living. She had intermittent brief, jerky movement of limbs of 2 weeks duration along with visual hallucination. No preceding febrile illness, head injury. No co-morbidities. At admission, the patient was conscious but with short attention span. Comprehension impaired. No eye movement restriction. Motor system examination revealed normal tone and deep tendon reflexes. In addition, there was startle myoclonus on examination. Gait was ataxic. Routine hematological and biochemical parameters including ammonia, vitamin B12 and thyroid functions were normal. Serum anti-TPO antibodies and ACE level were normal. ANA, VGKC receptor antibody was negative. Her serum ACE level was within normal limits. She was seronegative for HIV and serum VDRL. CSF analysis showed protein of 51 mg/dL and glucose of 61 mg/dL with 1 cell/cumm (lymphocytes). CSF gram stain, cryptococcal antigen, TB-PCR and HSV-DNA PCR were negative. EEG was abnormal, showing diffuse slowing of background rhythm (4.5 Hz) with bi-frontal, periodic sharp wave discharge occurring at 1 per second. The brain MRI showed T2/FLAIR hyperintensities in bilateral caudate with diffusion restriction in the same areas, diffusion hyperintensities in left parieto-occipital cortex. She was discharged and succumbed to illness within 8 weeks of admission. The patient was diagnosed as probable CJD in view of clinical symptoms of dementia, behavioral change, myoclonus, ataxia and visual hallucination, duration of disease less than 2 years, MRI and EEG abnormality.




