
Translate this page into:
Ewing's Sarcoma of the Cranial Vault
Address for correspondence: Dr. Geetha Narayanan, Department of Medical Oncology, Regional Cancer Centre, Trivandrum - 695 011, Kerala, India. E-mail: geenarayanan@yahoo.com
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Primary Ewing's sarcoma (EWS) arising from cranial bones is rare and accounts for only 1%–4% of all EWS. We report the case of a 15-year-old girl with EWS of the frontoparietal region of the skull. She underwent excision following which she received combination chemotherapy with vincristine, doxorubicin, cyclophosphamide alternating with ifosfamide, and VP16 and local radiation of 45 Gy. She is alive in complete remission at 40 months.
Keywords
Chemotherapy
Ewing's sarcoma
skull

INTRODUCTION
Ewing's sarcoma (EWS) accounts for 10% of malignant bone tumors and commonly arises from the long bones, pelvis, and ribs. Primary EWS arising from cranial bones is rare and accounts for only 1%–4% of all EWS.[1] We report the case of a 15-year-old girl with EWS of the skull and discuss the treatment of this rare entity.
CASE REPORT
A 15-year-old girl presented with swelling on the vertex of the head since 1 year, rapidly enlarging since few days. Computed tomogram (CT) showed an expansile lytic lesion with elevation of periosteum in the mid frontoparietal calvarium projecting into the scalp with heterogenous soft-tissue component [Figure 1]. The lesion was extending intracranially. A magnetic resonance (MR) angiogram showed lytic destructive mass lesion of frontal calvarium breaching the dural lining and partially encasing the superior sagittal sinus [Figure 2]. She did not have any sign or symptom of raised intracranial tension. She underwent wide excision of the mass and reported to us. Histopathological examination showed a highly cellular and vascular tumor composed of uniform round cells with scanty cytoplasm and round vesicular nucleus with periodic acid–Schiff + granules suggesting a malignant round cell neoplasm [Figure 3]. On immunohistochemistry, the cells showed strong membrane positivity for MIC 2 and were negative for leukocyte common antigen (LCA) and synaptophysin [Figure 4]. The picture was suggestive of EWS.
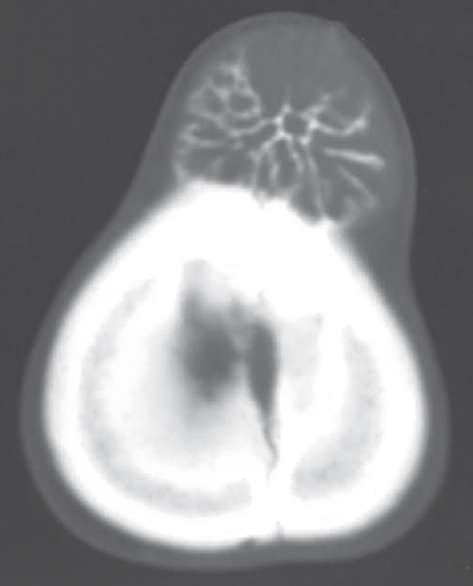
- Computed tomography scan showing the expansile lytic lesion in frontoparietal calvarium projecting into the scalp with soft-tissue component
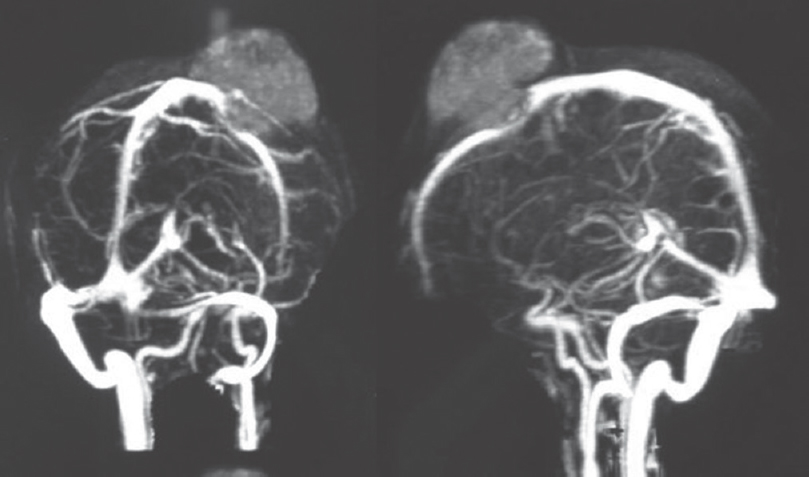
- Magnetic resonance angiogram showing the destructive mass lesion in frontoparietal calvarium
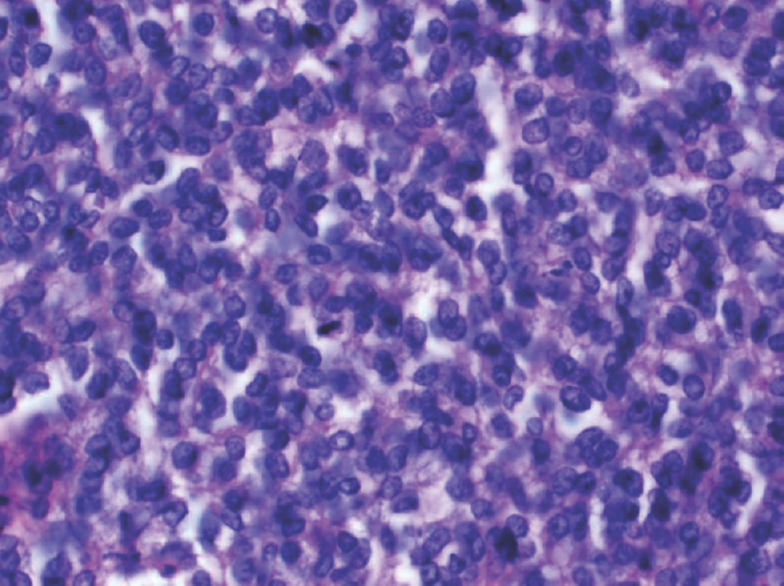
- Section showing cellular small round cell tumor with vesicular nuclei and vacuolated cytoplasm. A mitotic figure is seen toward the center of the field (H and E, ×400)
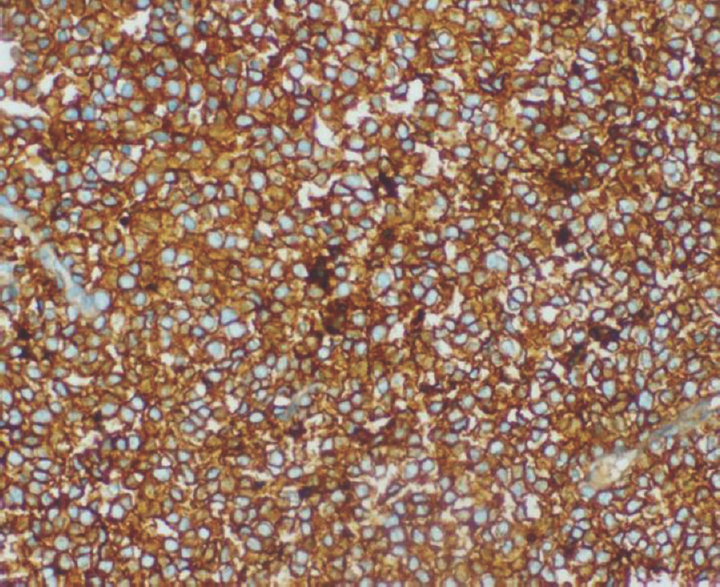
- Tumor cells showing diffuse and strong membrane staining for MIC 2 (×200)
Her hematology and serum chemistries were normal. A cerebrospinal fluid study, CT of the chest and bone marrow examination were normal. A technetium99 bone scan showed increased uptake peripherally with central cold area. She received combination chemotherapy with vincristine, doxorubicin, cyclophosphamide alternating with ifosfamide, and VP16 for 1 year. Local radiation of 45 Gy/25# was given at 9–12 weeks. She is alive in complete remission at 40 months.
DISCUSSION
EWS is a highly malignant bone tumor seen in children and young adults, and primary involvement of the skull is rare. EWS of the skull affects the frontal, parietal, and temporal bones and sometimes the skull base.[12] EWS typically grows extradurally and often reaches a large size before it is clinically detectable.[23]
In a retrospective study, among 332 cases of EWS, only seven were of the primary cranial involvement. The parieto-occipital region was the most common site.[4] About 90% of the cases occur in the first two decades of life, peak incidence is between 5 and 13 years of age, and males are affected more frequently.[2] Symptoms develop due to dural invasion, hydrocephalus, or raised intracranial pressure. The most common symptoms are headache and scalp swelling, the duration of which ranges from 2 weeks to 2 years.[15] Papilledema is seen in 80% of cases.[6] Our patient was 15 years, and she also had a similar presentation.
Plain X-ray of the skull shows a lytic lesion with mottling and erosion, and the classical onion peel appearance is rare. CT and MR imaging are useful to understand the size, extent, and involvement of dura and brain parenchyma. CT scan is useful to delineate bone involvement, and on MRI, the mass is hypo- to iso-intense on T1-weighted and iso- or hyper-intense on T2-weighted with heterogeneous enhancement on contrast.[4] On bone scintigraphy, an increased radioisotope uptake is seen.
Histologically, these tumors are characterized by sheets of small round blue cells with a high nuclear-cytoplasmic ratio.[125] Immunohistochemistry helps differentiate EWS from other small round cell tumors such as rhabdomyosarcoma, neuroblastoma, and lymphoma. EWS is positive for CD99 and vimentin and negative for desmin, LCA, and synaptophysin. Our patient also was positive for MIC 2 and negative for LCA and synaptophysin. EWS is characterized by a balanced translocation between chromosome 11 and 22 which fuses EWS gene on chromosome 22q12 with FLI1 gene on 11q24. Rarely, translocation t(21;22) or t(7;22) is observed. The fusion transcript type may determine the prognosis, with the presence of EWS–FLI1 fusion transcript type 1 being associated with improved outcome compared with that in patients with other fusion transcript types.[7]
EWS of cranium behaves differently from that in other areas in the following respects. The incidence of metastasis is less, radiologically the typical onion peel appearance may not be seen, and the prognosis is better than the disease in other sites.[6]
Like extracranial EWS, multimodal therapy is the treatment of choice for cranial EWS also. Local control can be achieved by surgery and/or radiation. Surgical resection is crucial in the management of EWS of the skull and is the preferred approach if the lesion is resectable. A radical excision should be attempted to minimize the tumor mass. In calvarial EWS, as majority of patients present with rapidly enlarging lesion and raised intracranial pressure, upfront surgical resection and decompression become imperative unlike EWS of extremities where presurgical chemotherapy is used to shrink the tumor.[6]
In calvarial EWS, it is usually not possible to get a margin free of microscopic disease and hence local radiotherapy is recommended at a dose of 40–50 Gy.[1]
Current chemotherapy regimen consisting of a combination of vincristine, cyclophosphamide, doxorubicin alternating with ifosfamide, and etoposide used for the treatment of EWS has given a 5-year survival rate of about 70%.[8] With total excision, radiation, and standard chemotherapy, the 5-year survival ranges from 39% to 65%.[12] Our patient underwent excision, received local radiation, and standard chemotherapy. She is alive in remission at 40 months.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- A case of primary Ewing's sarcoma of the occipital bone presenting with obstructive hydrocephalus. Childs Nerv Syst. 2003;19:792-9.
- [Google Scholar]
- Primary Ewing sarcoma of the petrous temporal bone: An exceptional cause of facial palsy and deafness in a nursling. Head Neck. 2006;28:955-9.
- [Google Scholar]
- Primary Ewing's sarcoma of the cranium: Case series and review of literature. J Cancer Res Ther. 2014;10:377-80.
- [Google Scholar]
- Primary Ewing's sarcoma of the occipital bone presenting as hydrocephalus and blindness. Pediatr Neurosurg. 2007;43:170-3.
- [Google Scholar]
- Primary Ewing's sarcoma of cranial bones: Analysis of ten patients. Acta Neurochir (Wien). 2011;153:1477-85.
- [Google Scholar]
- Ewing's sarcoma: Diagnostic, prognostic, and therapeutic implications of molecular abnormalities. J Clin Pathol. 2003;56:96-102.
- [Google Scholar]
- Addition of ifosfamide and etoposide to standard chemotherapy for Ewing's sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med. 2003;348:694-701.
- [Google Scholar]




