
Translate this page into:
Syndromes of Rapidly Progressive Cognitive Decline-Our Experience
Address for correspondence: Dr. Sadanandavalli Retnaswami Chandra, Faculty Block, Neurocentre, National Institute of Mental Health and Neurosciences, Bengaluru - 560 029, Karnataka, India. E-mail: drchandrasasi@yahoo.com
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Background:
Dementias are fairly slowly progressive degenerative diseases of brain for which treatment options are very less and carry a lot of burden on family and society. A small percentage of them are rapidly progressive and mostly carry a different course outcome. However, there are no definite criteria other than the time line for these patients.
Aims:
The aim of this was to identify and categorize the causes and course of rapidly progressive dementias seen in our center.
Settings and Design:
Patients who presented with rapid deterioration of cognitive functions within weeks to 1 year between 2011 and December 2016 were evaluated.
Patients and Methods:
All patients underwent all mandatory tests for dementia including brain imaging. Complete vasculitis workup, autoimmune encephalitis profile including Voltage Gated Potassium Channel, N-methyl-D-aspartic acid receptor, glutamic acid-decarboxylase, thyroid-peroxidase antibody, cerebrospinal fluid, and other special tests such as duodenal biopsy and paraneoplastic workup were done based on clinical indications.
Results and Conclusions:
Out of 144 patients 42 had immune-mediated encephalopathy, 18 had Creutzfeldt-Jakob disease, 3 had Vitamin B12 deficiency, 63 had infection with neurocysticercosis, 7 had tuberculosis, 2 had HIV, 1 had herpes simplex encephalitis, 1 had neurosyphilis, 1 Whipples disease, 1 had Subacute Sclerosing Panencephalitis, 1 had Mass lesion, 3 had Frontotemporal dementia, and 3 had small vessel disease. Good majority of these patients have infective and immune-mediated causes and less number belong to degenerative group. Therefore, caution is needed to look for treatable cause as it carries a different treatment options and outcome.
Keywords
Degenerations
immune-mediated
infections
nutritional
rapidly progressive dementia

INTRODUCTION
Unlike the usual course of dementias, certain types of dementias can develop over days, weeks, months, and rapidly end fatally, recover, or regress. They constitute a group of disorders which stem from diverse etiologies. However, if diagnosed properly, a good percentage of them can have a treatable cause. Therefore, they form a separate category.[1] For practical purpose, they can be grouped as prion- and nonprion-related disorders.[2] Among nonprion-related group, 39% still belong to the degenerative group which present and progress fast.[34] Third largest group belongs to unknown cause. The rest are immune mediated, infective, paraneoplastic, vasculitic, etc., There are variable reports regarding the frequency distribution of the various conditions in various studies. Antibodies against brain antigens are seen in paraneoplastic as well as nonparaneoplastic situations. Occasionally, toxins such as alcohol and other abused substances, metabolic disorders Like methylmalonic aciduria, late presentation of glutaric aciduria, mitochondrial cytopathies, and nutritional disorders such as B12 associated dementias show a rapid course. Making an early diagnosis is important and helps in segregating the treatable from the untreatable group and is of prognostic relevance for the ones who cannot be treated.
Prion disease and nonprion causes
Sporadic Creutzfeldt-Jakob disease (sCJD) usually presents as a cortico-striato-cerebellar syndrome with behavioral changes, delirium in persons between 50 and 70 years old with a mean survival of about 5 months. Vague nonspecific symptoms are also seen in a good number as malaise, weight loss, giddiness, etc. Probable sCJD was diagnosed with rapidly progressive dementia and at least two of the following clinical symptoms: pyramidal/extrapyramidal symptoms, visual or cerebellar disturbance, myoclonus, or akinetic mutism with typical electroencephalogram (EEG) changes or an increased cerebrospinal fluid (CSF) 14-3-3 protein level. Genetic variety of CJD has a slower course than the sporadic ones and affects younger people. (variant CJD); it often starts with psychiatric symptoms and abnormal movements including ataxia, myopathy, dementia, and paresthesias. Other variants are Heidenhain variant presenting with progressive visual disturbances and eventually blindness. Brownell-Oppenheimer variant begins with progressive cerebellar disturbance with unsteadiness and incoordination.
Alzheimer's disease (AD), diffuse Lewy body disease), corticobasal syndrome, and frontotemporal dementia (FTD) are also seen to show a fulminant course occasionally.[5] Whipple's disease is another condition which has a rapid course and needs a high degree of suspicion to diagnose. They present with dementia, oculo-facio-skeletal myorhythmia, new psychiatric symptoms, hypothalamic dysfunction, supranuclear gaze palsy, somnolence skin, and gastrointestinal symptoms. Hypothalamus, thalamus, and brainstem show hyperintensities and diagnosis is confirmed by PCR, brain biopsy, or duodenal biopsy; it is treatable with ceftriaxone 2 g daily for 28 days, doxycycline 100 mg twice daily, and hydroxychloroquine 200 mg three times daily.[6] Other infections such as HIV, tuberculosis (TB), and neurocysticercosis (NCC) manifest with dementia as a presenting symptom or secondary to vascular and other complications of the infections. However, there is no uniform criteria for diagnosis of the syndrome of rapidly progressive dementias, making diagnosis of conditions in this category a challenge.[7]
PATIENTS AND METHODS
Patients who presented to us with features of rapidly progressive cognitive decline in weeks to 1 year from January 2011 to December 2016 were evaluated. As, more than the time line and progressive cognitive decline, no other uniform criteria can be applied, they all underwent all mandatory tests for dementia including brain imaging. Complete vasculitis workup, autoimmune encephalitis profile including VGKC, N-methyl-D-aspartic acid receptor (NMDA), glutamic acid-decarboxylase (GAD), thyroid-peroxidase (TPO) antibody, CSF, and other tests such as duodenal biopsy were done based on clinical indications.
RESULTS
There were totally 144 patients seen by the authors. Forty-two patients had immune-mediated encephalopathy, 18 had CJD, 3 had Vitamin B12 deficiency, 63 had Infection with NCC, 7 had TB, 2 had HIV, 1 had Herpes simplex encephalitis, 1 had Neurosyphilis, 1 had Whipple, 1 had subacute sclerosing panencephalitis (SSPE), 1 had mass lesion, 3 had FTD, and 3 had small vessel disease (SVD). All patients had been thoroughly evaluated with detailed history and examination both neurological and systemic [Figure 1].
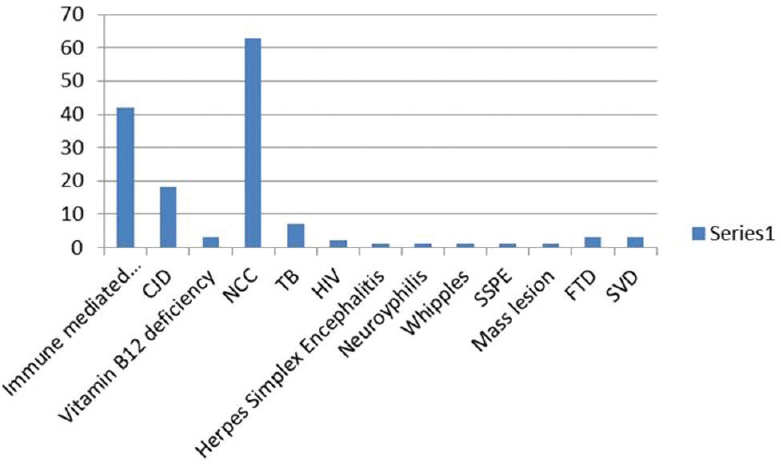
- Distribution of various diseases among patients with rapidly progressive dementia
DISCUSSION
These patient groups are categorized as prion related and nonprion related. The nonprion group is further characterized as patients with infective causes, immune mediated, and miscellaneous groups.
The prion-related cases
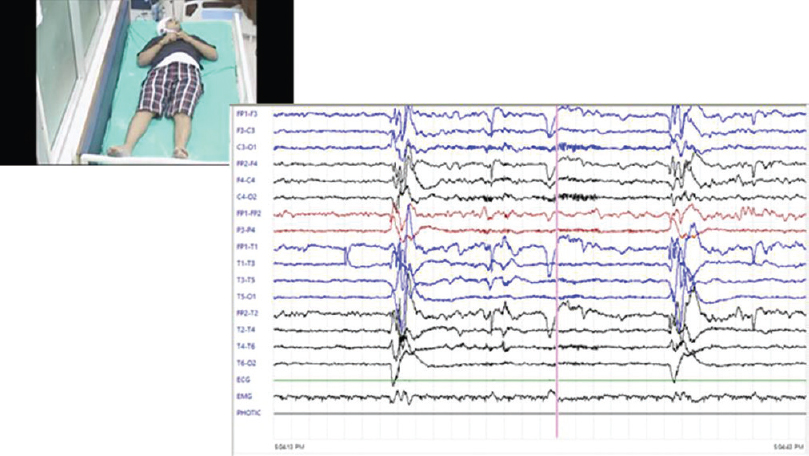
There were a total of 18 cases of probable CJD, of which two patients claimed family history of similar illness: one in mother and other in sister and both were females. One of the patients in the familial variety showed an unusual feature of trichotillomania [Figure 2]. One patient who presented as rapidly progressive blindness also had myokymia of shoulder, forearm, and chest muscles [Figure 3]. The diagnosis was made using clinical features, course, classical EEG changes seen in the sporadic forms, magnetic resonance imaging, and applying the revised WHO recommended standards and strategies for surveillance, prevention, and control of communicable diseases for probable CJD.[8] Eleven patients showed the typical EEG changes; others showed delta discharges. Males in the fifth to sixth decade are more commonly affected.[2] Diagnosis of this condition is important for prognostication and also plan precautions in handling of body fluids and tissues.[9]

- A case of familial Creutzfeldt-Jakob disease with trichotillomania
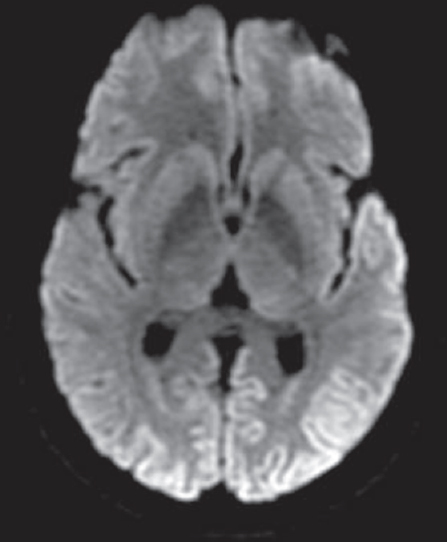
- Cortical ribboning in diffusion-weighted images of a patient with Creutzfeldt-Jakob disease
Infections
Among the nonprion group, the most common in this series was infection. The possibility of author bias cannot be excluded as the authors were specifically looking for such patients as part of another study. The most common infection we encountered presenting as rapidly progressive cognitive decline was neurocysticercosis. There were 63 patients diagnosed as definite or probable cases using Del Brutto et al.'s criteria.[10] More than two-third were male. Patients presented with encephalopathy, seizures, and raised intracranial tension and neuropsychological testing after the acute presenting symptoms were stabilized revealed predominant frontal and temporal dysfunction. These patients had majority of their lesions in the colloidal stage, had larger number of lesions, and showed increased levels of IL-10 and tumor necrosis factor alpha in CSF compared to controls. Acetylcholinesterase, which is present in the wall of this organism, was also elevated in CSF but was not statistically significant in the small proportion of 43 patients in whom it was done by us [Figure 4]. This enzyme was done as it can deplete acetylcholine and contribute to dementia.[11] Other infections were herpes simplex [Figure 5], neurosyphilis [Figure 6], SSPE, TB [Figure 7], and suspected Whipple in one patient [Figure 8 and Video 1] presenting as cognitive decline. The patient with neurosyphilis was being treated as degenerative versus central nervous system TB, but the typical trombone tongue gave us the clue and the diagnosis was later confirmed.
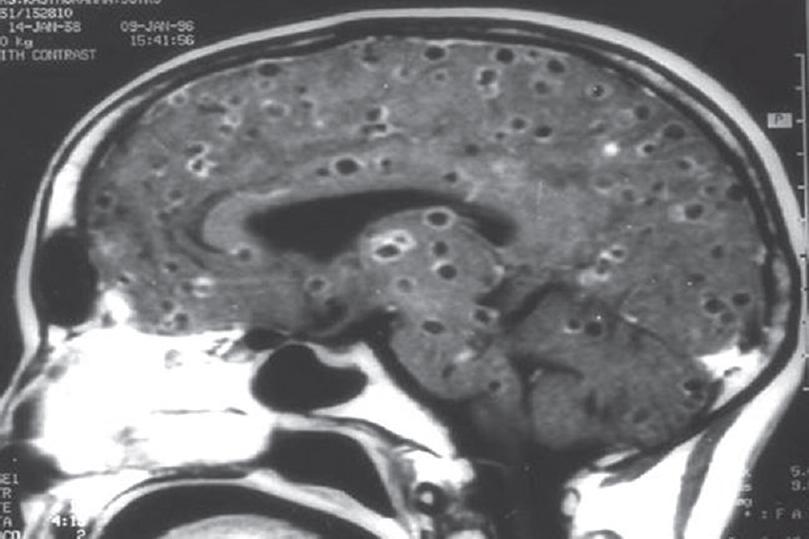
- Magnetic resonance imaging showing multiple neurocysticercosis
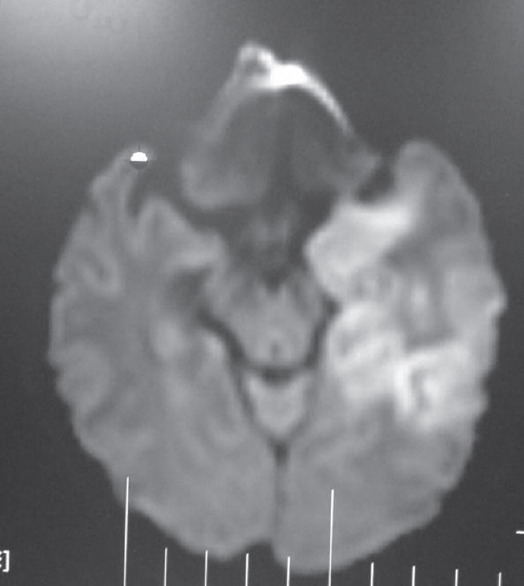
- T2 fluid-attenuated inversion recovery images showing hyperintensity in the temporal lobe left more than right in a patient with herpes simplex encephalitis
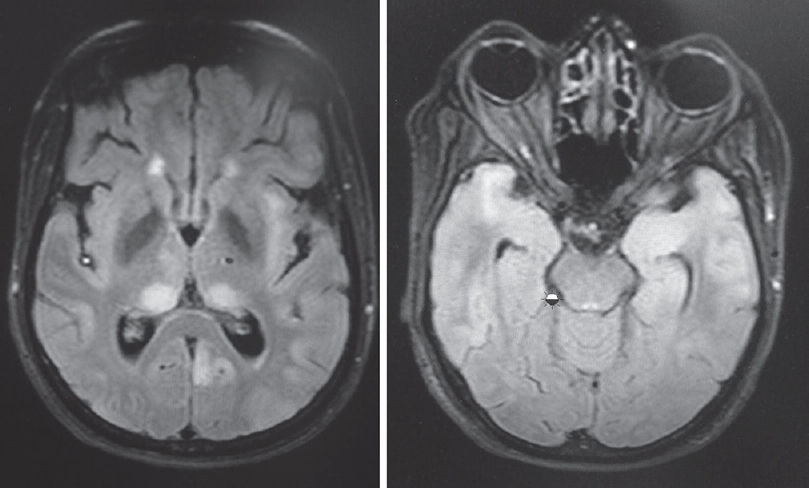
- T2 fluid-attenuated inversion recovery showing hyperintensities in thalamus, insula and both temporal lobes
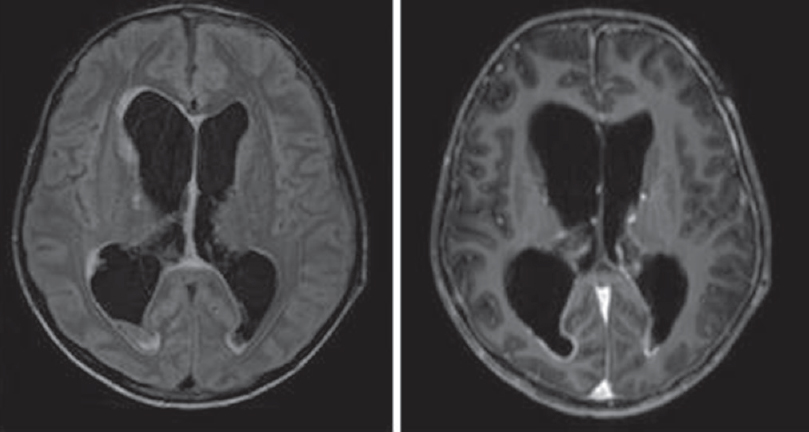
- Tuberculosis meningitis with hydrocephalus
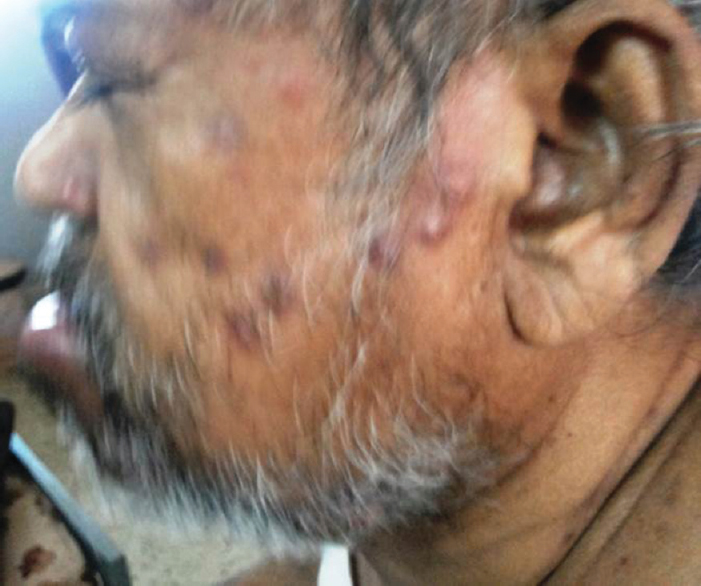
- Puckered skin lesions of Whipple disease
Neurotuberculosis
The most common presentation of TB in our setting is meningitis, hemiparesis, head ache, cranial neuropathy, etc. However, small percentage of patients manifested as rapid cognitive decline and they had hydrocephalous and multiple tuberculomas. One interesting observation in these patients was most of them were treated with quinolones as enteric fever which made the course slightly atypical and shifted to a subacute course.
Immune-mediated encephalopathy
The next largest group is immune-mediated encephalopathy which is a great imitator. Immune dysregulation associated with encephalopathies is being increasingly recognized as causally associated with a wide variety of rapidly progressive neuropsychiatric syndromes. They can be categorized into those associated with antibody to intracellular antigens such as anti-Hu seen with malignancy. Antibody to Extracellular epitopes of ion channels receptors and associated proteins like NMDA (N methy D Aspartate), Intracellular synaptic proteins like GAD65 (Glutamic Dehydraginase). There are Syndromes with less clearly established antigens like systemic lupus erythematosus. They present with neuropsychiatric syndromes with movement disorders, seizures, autonomic dysfunction, myoclonus, catatonia, dystonia, Isaac syndrome, ataxia, chorea, and peripheral neuropathy. A high degree of suspicion is needed to diagnose and treatment is lifesaving. We evaluated all suspected patients with rapidly progressive dementia were evaluated with CSF for viral infection including measles antibody titers and VGKC, NMDA, TPO antibody apart from all mandatory workup for patients with cognitive decline. A total of 42 patients were identified and their age varied from 11 years to 75 years. There were 13 females and 29 males.
First, 30 patients reported to a psychiatrist. Onset to diagnosis delay varied from weeks to 2 years. Their symptoms were unexplained anxiety in 69%, panic in 47%, uninhibited behavior in 14%, social incontinence in 12%, depression in 21%, psychotic features in 69%, unexplained paroxysmal symptoms of vague sensations in 2.1%, wandering tendency in 12%, myoclonic jerks in 43%, seizures in 50%, and stroke-like episodes in 7.1%. Papilledema was present in 4.4%, optic atrophy in 7%, mild hemiparesis in 7.1%, and ophthalmoplegia in 2.5%. Catatonia was seen in six patients,[12] paraneoplastic in two males with adenocarcinomatous deposit [Figure 9] and one female with carcinoma breast. Distribution of antibody was as follows: antinuclear antibody was positive in 6 cases of vasculitis (Figure 10 MRI in a case of vasculitis) and antibody to VGKC in 6 cases each combined VGKC and HIV in one patient, NMDA in eight patients, anti-Ri in one patient, TPO in three patients, negative in seven, and not done in eight patients. CSF measles antibody was significantly elevated in six patients resulting in a false diagnosis of SSPE in these patients.[13]
- Angiosarcoma scalp deposit with anti-Ri antibody positive dementia
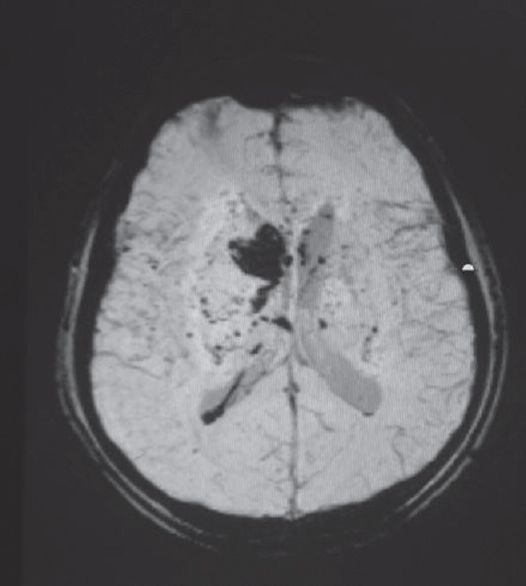
- Susceptibility weighted sequence of MRI showing small and large bleeds in a case of vasculitis
Whipple disease
Male patient aged 63 years presented with fever, weight loss, optic atrophy, neuropsychiatric symptoms, cognitive dysfunction, parkinsonian features, upgaze restriction, and oculomasticatory dysrhythmia and skin changes and following a protracted diarrheal illness after pilgrimage on Hajj. He also had typical skin changes [Figure 8]. Duodenal biopsy was done but was inconclusive. However, in view of the typical phenotype [Video 1], the patient was treated with ceftriaxone 2 g bd for 4 weeks followed by chloroquine 200 mg twice daily and sulfamethoxazole-trimethoprim. The patient showed significant improvement steadily over 6 months to 1 year. However, after 18 months, he passed away due to bronchopneumonia.
Other infections
Less commonly seen infection-associated dementia in this group are: HIV-associated dementia in one case, neurosyphilis associated dementia in one case, and late onset SSPE presenting as dementia in one case [Figure 11].
- Subacute sclerosing panencephalitis patient with classical periodic complexes
Frontal lobe glioma
A 70-year-old female patient admitted with progressive frontotemporal syndrome with no features of raised intracranial pressure was diagnosed as large glioma of frontal lobe [Figure 12].
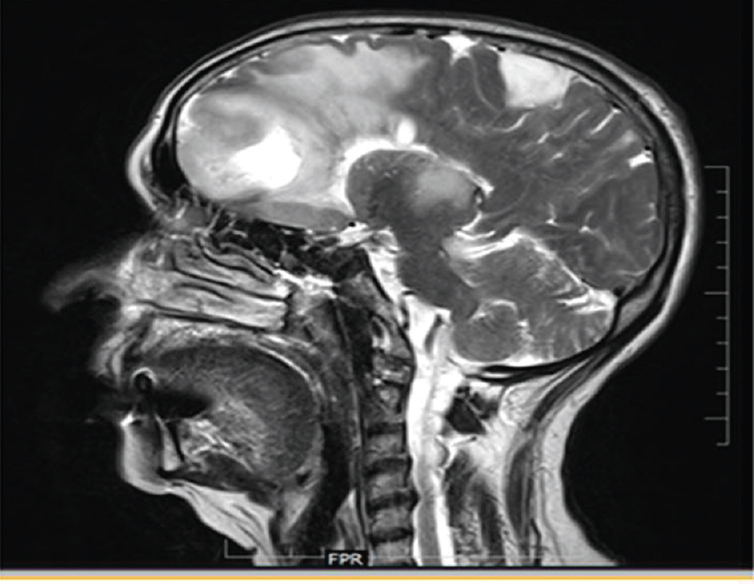
- Sagittal brain section showing a large frontal mixed density lesion with a satellite lesion in the posterior frontoparietal region
Neurodegenerative dementia
Among our patients with rapidly progressive cognitive decline, there were three patients each with FTD and SVD.
Nutritional
Three of our patients with rapidly progressive dementia had severe B12 deficiency and atrophic gastritis on gastric mucosal biopsy and recovered fully with B12 replacement.
CONCLUSIONS
Rapidly progressive cognitive decline is commonly seen in a wide spectrum of conditions varying from vascular, immune mediated, toxic, infective, metabolic, neoplastic, degenerative, drug related, as well as nutritional and degenerative conditions. Our study of 144 patients inthe last 5 years shows infections as the most common cause and immune mediated as the second commonest in our setting. As they are potentially treatable and partly or completely reversible based on the time of diagnosis, careful evaluation looking for a treatable cause is essential in these patients. Commonly, they present with neuropsychiatric symptoms resulting in significant delay in diagnosis. Screening for malignancy is indicated in elderly patients and those with NMDA antibody positivity and may have to be repeated in some patients. When cognitive decline is fast, common conditions such as infections should be looked for carefully. Thorough etiological workup is mandatory. CJD and autoimmune disorders can be great imitators. EEG can serve as good screening tool to differentiate organic from psychiatric disorders. In our study, a probable bias toward infections cannot be ruled out as the authors were engaged in analysis of infections.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Video Available on: www.ruralneuropractice.com
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgment
We acknowledge with gratitude National Institute of Mental Health and Neurosciences for the opportunity given to us to study these patients.
REFERENCES
- Creutzfeldt-Jakob disease phenotype and course: Our experience from a tertiary center. Indian J Psychol Med. 2016;38:438-42.
- [Google Scholar]
- Frontotemporal dementia progresses to death faster than Alzheimer disease. Neurology. 2005;65:719-25.
- [Google Scholar]
- Dementia with Lewy bodies. Contemporary Neurobehavioral Syndromes. Joachim, Morris: Selkoe; 2011. p. :111-29.
- Differential diagnosis of 201 possible Creutzfeldt-Jakob disease patients. J Neurol. 2004;251:298-304.
- [Google Scholar]
- Whipple's disease: A review of 19 patients from one hospital and a review of the literature since 1950. Medicine (Baltimore). 1970;49:175-205.
- [Google Scholar]
- The prion diseases. In: Seminars in Neurology. Vol 20. New York, USA: Copyright© 2000 by Thieme Medical Publishers, Inc; 2000. p. :337-52.
- Guidelines for high risk autopsy cases: Special precautions for Creutzfeldt-Jakob disease. In: Autopsy Performance and Reporting. Northfield, Illinois: College of American Pathologists; 1990. p. :68-74.
- [Google Scholar]
- Cognitive dysfunction and its determinants in patients with neurocysticercosis. Indian J Psychol Med. 2016;38:142-6.
- [Google Scholar]
- Catatonia in children following systemic illness. Indian J Psychol Med. 2015;37:413-8.
- [Google Scholar]
- Elevated antimeasles antibody titre: An association in autoimmune encephalitis. J Neurosci Rural Pract. 2015;6:536-40.
- [Google Scholar]




