
Translate this page into:
Supratentorial primitive neuroectodermal tumor presenting with intracranial hemorrhage in adult
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
A 24-year-old female patient presented with complaints of nausea, vomiting and of loss of consciousness lasted for 15 minutes with left sided weakness. Neuroradiological evaluation revealed a hemorrhagic mass lesion in the right frontal lobe. The patient was operated and intraoperative findings showed a cortical-subcortical hematoma including hemorrhagic and disrupted tissue with a pathologic purple tissue on the periphery of the hematoma. Postoperative course was uneventful and postoperative histopathological examination revealed primitive neuroectodermal tumor. The patient was then referred to medical and radiation oncology clinics for further evaluation and treatment.
Keywords
Brain tumor
intracranial hemorrhage
primitive neuroectodermal tumor

Introduction
Primitive neuroectodermal tumors (PNETs) are a group of highly malignant tumors composed of small round cells of neuroectodermal origin. Cranial PNETs are commonly located infratentorially in the cerebellum and are rarely supratentorial. These tumors are common primary central nervous system tumors in children, but they are extremely rare in adults, representing less than 0.5% of all intracranial tumors.[1] Although they are histologically very similar to medulloblastoma, supratentorial PNETs have markedly different clinical behavior and are generally considered to represent a more aggressive tumor group than medulloblastoma, with a frequently massive tumor burden and a higher incidence of disseminated disease at diagnosis.[2] PNETs in adults usually present with signs of increased intracranial pressure (ICP), seizures and/or focal neurological deficits.[1] However, supratentorial PNET presenting with intracerebral hemorrhage is very rare and only few similar reports are present.[3]
This report presents an unusual case of supratentorial PNET presented with intracranial hemorrhage in an adult patient. Clinical features and intraoperative and pathologic findings are discussed.
Case Report
A 24-year-old female patient was admitted to our emergency service with complaints of nausea, vomiting and loss of consciousness lasted for 15 minutes with left sided weakness. A neurological examination revealed left hemiparesis with motor power of 2/5 for both left arm and leg. The patient had no significant medical history. The chest X-ray, blood chemistry, and complete blood count were normal. A brain computed tomography (CT) scan showed a hyperdense mass lesion in the right frontal lobe consisting of hematoma with surrounding edema causing mass effect and midline shift [Figure 1a]. The patient was admitted to the intensive care unit (ICU) for follow-up and dexamethasone and prophylactic antiepileptic course was started. The patient showed improvement and the motor power of the left side returned to normal. Brain magnetic resonance imaging (MRI) revealed a 3-cm heterogeneous round the mass lesion in the right frontal lobe, anterior to and compressing the precentral gyrus. This mass was heterogeneous on T1 and T2 weighted images with irregular enhancement after contrast admission [Figures 1b–d]. Magnetic resonance arteriogram (MRA) and venogram (MRV) was done showing no evidence of vascular pathology. The patient was operated with the preliminary diagnosis of hemorrhagic brain tumor in the supine position with right fronto-parietal horseshoe incision and frontoparietal craniotomy. Intraoperative findings showed a cortical-subcortical hematoma including hemorrhagic and disrupted tissue with pathologic purple tissue on the periphery of the hematoma. The tumor did not express a good cleavage plan and resection continued until normal brain tissue was seen. Operation was ended without any complications. The postoperative period was uneventful, with no complications or neurologic deficits. Postoperative brain CT scan showed total removal of the tumor and hematoma [Figure 1e]. Histopathologic examination revealed the brain tissue with reactive gliosis, and mixed inflammatory cells infiltrate mainly neutrophils, hemorrhage and necrosis are also seen. The brain tissue was infiltrated by a poorly differentiated patternless tumor composed of uniform neoplastic medium-sized cells, with round regular nuclei and high nucleus: Cytoplasmic ratio, with delicate lobular fibrous stroma, Homer-Wright rosettes and vascular endothelial proliferation. Immunohistochemical staining showed positivity to vimentin, synaptophysin, glial-fibrillary acidic protein (GFAP) and cluster of differentiation 56 (CD56), but were negative for desmin, epithelial membrane antigen (EMA) and factor 8, with high Ki-67 index indicating increased mitotic activity and a high-grade malignancy. The resected lesion was identified as a central nervous system grade IV PNET [Figure 2]. Chromosomal analysis showed distal chromosome 4q loss. The patient was discharged and referred to medical and radiation oncology clinics for further evaluation and treatment. Chest, abdomen and pelvic CT-scans were normal. Spinal MRI with contrast did not show any sign of tumor seeding. Cerebrospinal fluid CSF cytology revealed no pathologic cells. However, radiotherapy was planned to include the spinal axis.
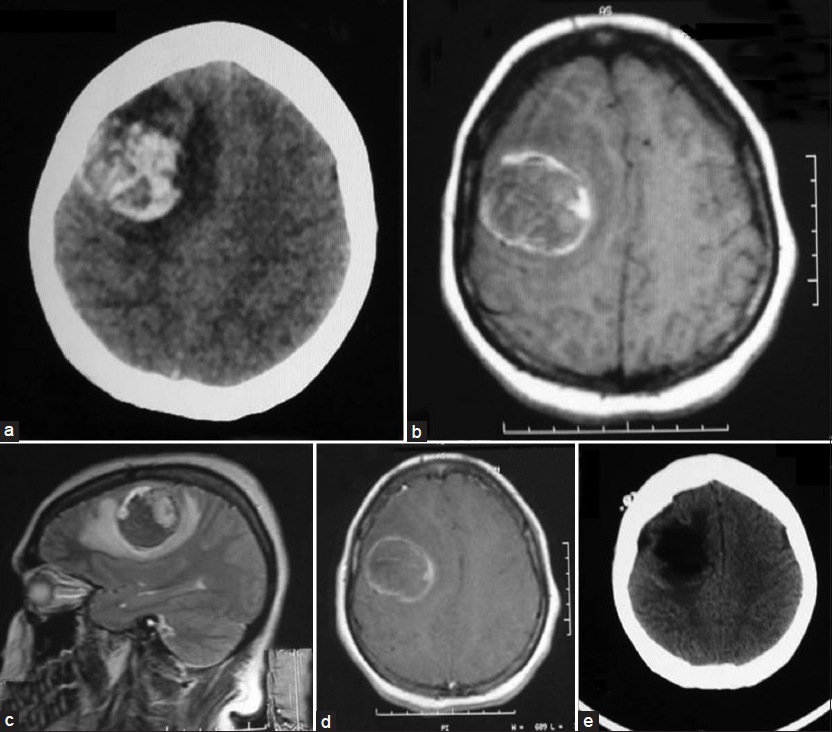
- Preoperative neuroimaging of the present case showing the right frontal hemorrhagic mass. (a) Brain CT scan, (b) Axial T1-weighted MRI, (c) Sagittal T2-weighted MRI, (d) Axial post-contrast enhancement MRI, (e) Post-operative brain CT scan demonstrating total resection of the mass and hematoma
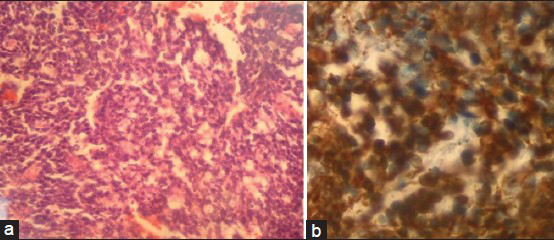
- (a) A photomicrograph demonstrating small tumor cells with round to oval hyperchromatic nuclei and scant cytoplasm (×100 hematoxylin and eosin), (b) Photomicrograph showing intense positivity for synaptophysin (×400 immunohistochemical stain)
Discussion
PNET's comprise a heterogeneous group of malignant tumors that mainly affect the pediatric population. These tumors composed of neuroepithelial cells which are able to differentiate along the neuronal, astrocytic, muscular, or melanocytic lines, with high index of proliferation.[3] The term PNET was introduced in 1973 for describing initially supratentorial, pediatric brain tumors that were largely undifferentiated or primitive but contained small foci of glial or neuroblastic differentiated cells of primitive neuroectodermal origin.[4] PNETs of the central nervous system include neuroblastomas, ganglioneuroblastomas, medulloepitheliomas, ependymoblastomas, embryonal tumors with abundant neuropil and true rosettes, and not otherwise specified PNET tumors.[3] The previous long-standing controversy regarding the inclusion of medulloblastomas and pineoblastomas into the PNET category has been resolved by the WHO tumor classification.[3] Supratentorial PNETs are very rare and aggressive tumors, which corresponds to WHO grade IV, and account for approximately 2.5%-3% of all pediatric tumors.[2] These tumors are even more rare in adult the population representing less than 0.5% of all intracranial tumors.[1]
Most patients present with symptoms and signs of raised ICP such as headaches, nausea and/or vomiting, confusion and papilledema because of the rapid tumor growth.[1] Furthermore, an adult supratentorial PNET may present with focal neurological signs and deficits, such as limb paresis, aphasia, facial palsy and visual field defects, depending on the anatomic location of the tumor.[1] Occasionally, the clinical presentation may involve seizures.[1] However, supratentorial PNET presenting with intracerebral hemorrhage is very rare and only few similar reports are present.[5] Because of the small numbers of individual cases, no study has shown the exact incidence of hemorrhagic presentation.
Supratentorial PNETs on CT scan are isodense or hyperdense.[1] Moreover, the locations of these tumors are almost equally distributed in the frontal, temporal, and parietal lobes. On MRI, these lesions appear hypo and hyperintense on T1 and T2-weighted sequences, respectively and in most of the cases there is heterogeneous contrast enhancement after gadolinium administration.[1] Some areas of decreased diffusion within the lesion are common findings, due to the high cell density. Also, there may be intratumoral hemorrhage, cysts, calcification, surrounding brain edema, and necrosis.[1] Proton magnetic resonance spectroscopic studies have shown nonspecific increased choline and lactate, and decreased N-acetyl-aspartate concentrations.[1] In our case, the tumor presented as intracranial hemorrhage with classical finding of hematoma in CT scan and MRI. Differential diagnosis of our case included mainly vascular etiologies, such as, cavernoma, arteriovenous malformation (AVM), arteriovenous fistula (AVF), hemorrhagic venous infarct.
Grossly, adult PNETs usually appear as lobulated, purple-grayish or pinkish masses. On microscopic examination, the tumor cell population consists of undifferentiated small cells with ill-defined, scanty cytoplasm, and round or oval cells with hyperchromatic nuclei.[6] Mitotic activity is variable. Microscopic calcifications, necroses and Homer-Wright rosettes are also observed in a number of cases. The vascularity of the tumors varies, whereas endothelial cell proliferation within the vessel wall is regularly observed.[6]
PNET's are considered a challenging clinical entity for neurosurgeons due to the poor prognosis it confers. When compared with high risk medulloblastoma patients treated with the same regimens, patients with supratentorial PNETs fare worse in terms of survival rates, suggesting that there is an intrinsic difference in the biology of these tumors.[7] The overall 5-year progression-free survival rate for patients who do not undergo resection ranges from 18% to 47%.[2] The reported overall survival of PNETs in the pediatric population ranges between 29% and 57%.[6] Moreover, the prognosis for adults with supratentorial PNETs seems to be worse than that of their pediatric counterparts.[16] Most adults, with limited exceptions, with a supratentorial PNET die within a year of the initial diagnosis. There are a few reports on local recurrence and spinal and lung metastases.[5] For pediatric patients with supratentorial PNETs, the prognosis correlates with age, extent of necrosis, tumor dissemination and the presence of bad prognostic genetic features, such as, MYCN or MYCC gene amplifications and polysomies of chromosomes 2 and 8.[16] By contrast, in adults the most important prognostic factor is considered to be the Ki-67 index. Kim et al.[1] Reported that adult patients with a Ki-67 index greater than 30% demonstrated very poor outcome, with a mean postoperative survival time of eight months.
When possible, the complete resection along with radiation and chemotherapy has been shown to lead to better survival in several studies.[2] The extent of resection depends on several factors and must be weighed against the risks of postoperative neurological deficits. Supratentorial PNETs occurring in eloquent areas of the brain presents a particularly difficult situation because the risks of severe postoperative neurological deficit are relatively high. These tumors have a high tendency for spinal metastasis and approximately 30-50% of the patients with intracranial PNETs develop spinal metastases.[8] Postsurgical irradiation of the whole craniospinal axis may be employed, even when there is no evidence of spinal seeding.[1] The employed mean dose is 54Gy for the brain and 31Gy for the spinal cord.[1] The exact role of stereotactic radiosurgery employment, especially in cases of partially resected PNETs, remains to be defined.
Source of Support: Nil.
Conflict of Interest: None declared.
References
- Supratentorial primitive neuroectodermal tumors in adults. J Neurooncol. 2002;60:43-52.
- [Google Scholar]
- Pineal and nonpineal supratentorial primitive neuroectodermal tumors. Childs Nerv Syst. 1999;15:586-91.
- [Google Scholar]
- The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114:97-109.
- [Google Scholar]
- A supratentorial primitive neuroectodermal tumor presenting with intracranial hemorrhage in a 42-year-old man: A case report and review of the literature. J Med Case Rep. 2013;7:86.
- [Google Scholar]
- Central nervous system primitive neuroectodermal tumors: A clinicopathologic and genetic study of 33 cases. Brain Pathol. 2010;20:441-50.
- [Google Scholar]
- Patterns of failure in supratentorial primitive neuroectodermal tumors treated in Children's Cancer Group Study 921, a phase III combined modality study. Int J Radiat Oncol Biol Phys. 2004;60:204-13.
- [Google Scholar]
- Intracranial metastasis from primary spinal primitive neuroectodermal tumor. Asian J Neurosurg. 2013;8:42-7.
- [Google Scholar]




