
Translate this page into:
Spinal Muscular Atrophy and Progressive Myoclonic Epilepsy: A Rare Association
Niraj Kumar, MD, DM Department of Neurology, All India Institute of Medical Sciences Rishikesh 249201, Uttarakhand India drnirajkumarsingh@gmail.com
This article was originally published by Thieme Medical and Scientific Publishers Pvt. Ltd. and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Abstract
The association of spinal muscular atrophy (SMA) with progressive myoclonic epilepsy, also known as “SMA plus,” is a unique syndrome linked to non-survival motor neuron (non-SMN) genes. The disease starts in childhood with progressive weakness and atrophy of muscles; myoclonic epilepsy develops during later childhood, after the onset of motor symptoms. In this report, we describe a case of SMN gene unrelated SMA and myoclonic epilepsy, supported by electrophysiological and neuropathological evidences.
Keywords
SMA
PME
SMA-PME
SMA plus
spinal muscular atrophy
progressive myoclonic epilepsy

Introduction
Progressive lower motor neuron (LMN) type weakness and diffuse muscle atrophy characterize spinal muscular atrophy (SMA). An autosomal recessive disorder by inheritance, SMA is caused by the loss of ventral horn cells in the spinal cord and cranial nerve nuclei in the lower brainstem. Most of SMA cases are linked to survival motor neuron (SMN) gene.1 Sometimes patients with SMA can have atypical features like myoclonic epilepsy (“SMA Plus”) unlinked to SMN gene. Mutations in certain other genes like N-acylsphingosine amidohydrolase1 (ASAH1) may be seen in such cases.2 We report a case of a young boy presenting with SMA and myoclonic epilepsy, unrelated to SMN gene.
Case Presentation
A 6-year-old boy, product of nonconsanguineous marriage, presented with insidious onset of gradually progressive symmetrical upper and lower limb weakness since the age of 2 years. There was no history of perinatal insult. All his motor milestones were delayed and he was never able to run or climb stairs. Weakness was more in the proximal group of muscles. He developed neck weakness along with thinning of both arms and thighs by age 3. At 3.5 years of age, he developed sudden brief jerky movements involving extremities followed by fall. At 4 years of age, he started having difficulty in speaking and swallowing with occasional episodes of nasal regurgitation. Weakness and muscle wasting progressed and 3 years into illness he developed truncal weakness. Frequency of the jerks increased with time; his parents noticed postural tremors and twitches involving both the hands. There were brief episodes of staring look and involuntary eye blinking associated with impaired awareness. There was no sensory complaints, cognitive impairment, ataxia, or bladder bowel involvement. Family history was noncontributory. Examination showed diffuse muscle wasting along with LMN type of weakness involving both upper and lower limbs. Deep tendon reflexes were attenuated globally. Gag reflex was poor and tongue was atrophied with florid fasciculations. Postural tremor was observed in both upper limbs along with polyminimyoclonus. Myoclonic jerks involving axial and appendicular musculature were seen.
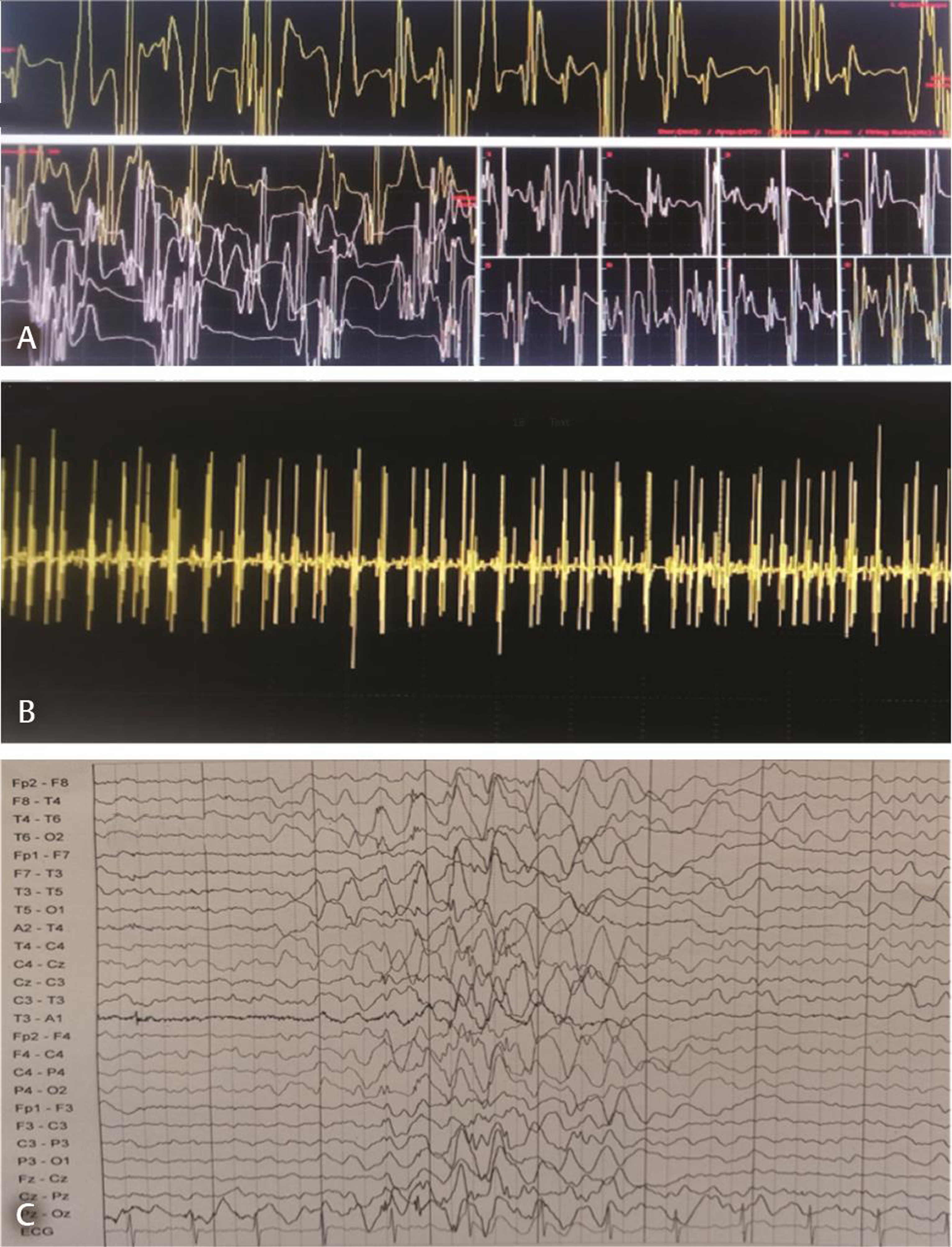
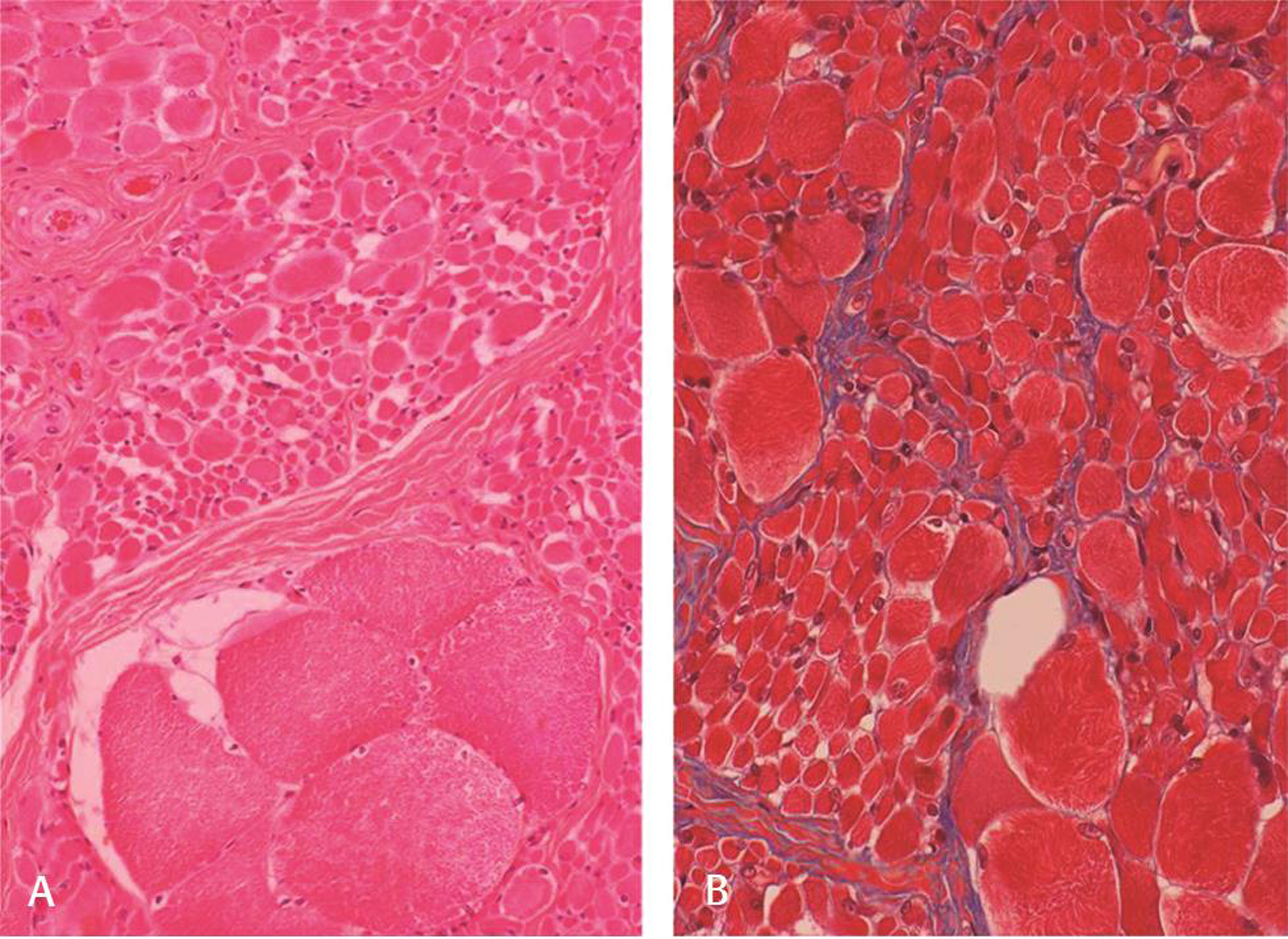
Nerve conduction study was normal; needle electromyogram revealed prominent fibrillation potentials, positive sharp waves, and fasciculations in paraspinal muscles and sampled muscles of extremities. The motor unit action potentials were of large amplitude with polyphasia, prolonged duration, and reduced interference pattern (Fig. 1A B). Electrophysiological features suggested diffuse anterior horn cell disease. Magnetic resonance image of the brain and spine was normal. Electroencephalogram (EEG) showed generalized polyspike and wave (2.5–4 Hz) epileptiform discharges (Fig. 1C). The left quadriceps biopsy showed fascicles with groups of hypertrophied rounded fibers and small atrophic fibers suggestive of neurogenic changes (Fig. 2A B). No deletions or mutation was found in SMN gene. Skin biopsy done to exclude Lafora body disease was noncontributory. Clinical, laboratory, and histopathological findings ruled out the possibility of mitochondrial disease. Genetic tests for ASAH1,mutation, hexosamindase A levels, or tests for lysosomal storage disorders were done due to financial constraints. Myoclonic jerks and EEG findings responded to treatment with valproic acid and clobazam.
-
Fig. 1 Electromyogram showing motor unit action potentials of large amplitude with long duration, polyphasia (A), and reduced interference pattern on maximum voluntary contraction (B), with following setting: 100 uV/10 ms; 3 kHz/50 Hz. Electroencephalogram reveals generalized polyspike and wave (2.5–4 Hz) epileptiform discharges (C).
Fig. 1 Electromyogram showing motor unit action potentials of large amplitude with long duration, polyphasia (A), and reduced interference pattern on maximum voluntary contraction (B), with following setting: 100 uV/10 ms; 3 kHz/50 Hz. Electroencephalogram reveals generalized polyspike and wave (2.5–4 Hz) epileptiform discharges (C).
-
Fig. 2 Muscle biopsy shows fascicles with groups of hypertrophied rounded fibers and small atrophic fibers suggestive of spinal muscular atrophy (A) hematoxylin and eosin x Obj. 10; (B) Masson trichrome x Obj. 20. Note absence of endoneurial fibrosis (B).
Fig. 2 Muscle biopsy shows fascicles with groups of hypertrophied rounded fibers and small atrophic fibers suggestive of spinal muscular atrophy (A) hematoxylin and eosin x Obj. 10; (B) Masson trichrome x Obj. 20. Note absence of endoneurial fibrosis (B).
Discussion
The variants of SMA with atypical clinical features (SMA plus syndromes) reported in literature are not linked to SMN gene.3 Some patients with spinal muscular atrophy–progressive myoclonic epilepsy (SMA-PME) phenotype carry mutations in ASAH1.2 Jankovic and Rivera first identified association of SMA with PME in 1978.4 But genetic tests were not available at that time. The first report of SMA associated with PME linked to ASAH1 was published in 2012.2 To best of our knowledge, 44 cases of SMA-PME-like clinical presentation have been reported to date and 24 of them carried ASAH1 mutation.2 4 5 6
Muscle weakness and wasting along with myoclonic jerks and seizures were common in all the reported cases of SMA-PME despite the differences in attributes like age of onset, severity and progression of the disease. Less consistent features reported include tremors, cognitive impairment, sensorineural hearing loss, atrophy of cerebral and cerebellar hemispheres. Disease onset is characterized by muscle wasting and weakness during early childhood and epilepsy develops later, after the onset of motor symptoms. ASAH1 mutations are also linked to a more severe form of disease called Farber lipogranulomatosis (FL).5 However, joint and skin findings of FL are not reported in SMA-PME.2 3 4 5
Until now no case of SMA-PME phenotype has been reported from the Indian subcontinent. In this report we describe a case of a young boy having SMA associated with unique clinical and electrophysiological features of myoclonic epilepsy. Review of literature indicates that SMA with myoclonic epilepsy, unrelated to SMN gene, is a unique condition with its own clinical and electrophysiological features.Study of additional cases is also required to understand the genetic and pathophysiological basis of SMA-PME.
Acknowledgments
The authors would like to express their gratitude to the patients and their family.
Conflict of Interest
None declared.
Funding None.
References
- Diseases of the motor unit.Child Neurology. (6th edition). Philadelphia: Lippincott Williams and Wilkins; 2000. p. :1027-1092. In: eds.
- [Google Scholar]
- Spinal muscular atrophy associated with progressive myoclonic epilepsy is caused by mutations in ASAH1. Am J Hum Genet. 2012;91(1):5-14.
- [Google Scholar]
- 93rd ENMC International Workshop: non-5q-spinal muscular atrophies (SMA) - clinical picture (6-8 April 2001, Naarden, The Netherlands) Neuromuscul Disord. 2003;13(2):179-183.
- [Google Scholar]
- Hereditary myoclonus and progressive muscular atrophy: a new syndrome. Trans Am Neurol Assoc. 1978;103:116-118.
- [Google Scholar]
- Acid ceramidase deficiency: Farber disease and SMA-PME. Orphanet J Rare Dis. 2018;13(1):121.
- [CrossRef] [Google Scholar]
- A novel case report of spinal muscular atrophy with progressive myoclonic epilepsy from Iran. Int Med Case Rep J. 2019;12:155-159.
- [Google Scholar]




