
Translate this page into:
Parasellar Chondrosarcoma in Three Young Patients: A Diagnosis of Caution
Address for correspondence: Prof. Ravindra Kumar Saran, Department of Pathology, GB Pant Institute of Postgraduate Medical Education and Research, New Delhi - 110 002, India. E-mail: ravindraksaran@hotmail.com
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Parasellar chondrosarcoma is rare slowly growing intracranial tumors. A correct diagnosis of these tumors is challenging for clinician due to overlapping location and simulation of clinical presentations with much common pituitary adenomas. We are reporting three young patients diagnosed to have parasellar chondrosarcoma highlighting the pathological features of importance required for confirm diagnosis.
Keywords
Chondrosarcoma
chordoma
parasellar

INTRODUCTION
Pituitary adenoma is the most common sellar mass. About 10% of the sellar/parasellar tumors are of nonpituitary origin.[1] It is very challenging to differentiate these lesions radiologically from the much common adenomatous tumors due to their common location. Parasellar chondrosarcomas are nonpituitary cartilaginous tumor of the sellar region which arises from skull base and accounts for 0.15% all intracranial tumors.[2] Herein, we are describing three cases of parasellar chondrosarcomas emphasizing the pathological features of importance for accurate diagnosis.
CASE REPORTS
Case 1
A 29-year-old male patient presented with the chief complaints of headache on and off for 6 months and deviation of the left eyeball, ptosis along with diminution of vision for 3 months. Magnetic resonance imaging (MRI) of the brain revealed a large multilobulated extra-axial skull base lesion in the parasellar region arising from the left lateral wall of sphenoid and left petroclival synchondrosis appearing hypointense on T1W1 and bright hyperintense on T2W1 with intralesional calcification, postcontrast enhancement, and mass effect [Figure 1].
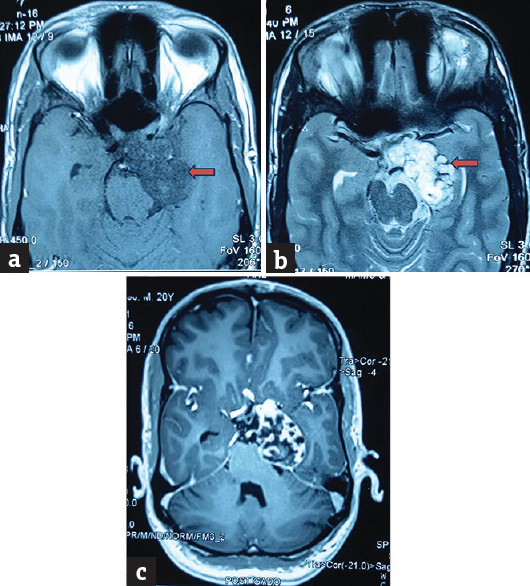
- Magnetic resonance imaging showing (a) a large multilobulated extra-axial skull base lesion in the parasellar region arising from the left lateral wall of sphenoid and left petroclival synchondrosis appearing hypointense on T1W1, (b) bright hyperintense on T2W1 with intralesional calcification and (c) postcontrast enhancement
Left frontotemporal craniotomy was done. Intraoperatively dura was tense, and the brain was bulging, left anterior temporal lobectomy done to reach the tumor through subtemporal intradural approach. Tumor was of mixed variety stony hard at some places and soft at other places, moderately vascular, and encapsulating petrous portion of the left internal carotid artery (ICA).
Case 2
A 22-year-old male presented with the chief complaints of headache for 1 year and diminution of vision, diplopia, and restriction of movement in the left eye for the past 3 months. Contrast-enhanced computer tomography showed irregular calcified lesion in left parasellar and suprasellar region measuring 27 mm × 23 mm with minimal enhancement [Figure 2]. A radiological diagnosis of craniopharyngioma was made. Left parietal craniotomy and tumor decompression were done. Intraoperatively tumor was hard, pinkish white, moderately vascular and engulfing the left ICA.

- (a) Axial computer tomography images at the level of sella showing expansile lobulated lesion with calcified matrix in the left parasellar location (involving petroclival synchondrosis), (b) axial fluid-attenuated inversion recovery showing heterogeneous signal intensity with multiple hyperintense spaces pointing towards chondroid origin, (c) axial postcontrast, and (d) sagittal postcontrast images showing heterogeneous enhancement
Case 3
A 19-year-old female presented with the chief complaints of on and off throbbing headache localized to the left side for 1 month. MRI of the brain revealed basifrontal bilateral extra-axial mass lesions with significant perilesional edema suggestive of meningioma. Bifrontal craniotomy and tumor decompression were planned. Intraoperatively tumor was firm, nonsuckable, moderately vascular with well-defined planes and attached to falx.
DISCUSSION
About 10% of the sellar/parasellar lesions are nonpituitary in origin, of which 10% are cartilaginous include both chordoma and chondrosarcoma. They are thought to arise from the remnants of embryonic cartilage tissue in the cartilaginous junction of cranial base sutures.[3] These are the rare slowly progressing tumors usually present with headache and visual disturbances. Due to the complexity of their location, cranial nerves, and major vascular structures, complete removal is sometimes very difficult and results in recurrence.
Chondrosarcomas are malignant tumors of cartilage-forming cells which rarely may be associated with Ollier disease, Maffucci syndrome, and Paget's disease. The different histological subtypes of chondrosarcoma include conventional, mesenchymal, clear cell, and dedifferentiated subtypes.[4] Chondrosarcomas are isointense to hypointense on T1 and overall hyperintense on T2 with foci of low T2 signal corresponding to calcified chondroid matrix.[5]
Histologically, the tumor showed well-defined lobules of hyaline cartilage along with trabecular and lamellar bone. The tumor comprised bland looking chondrocytes with focal cellular atypia and hypercellularity. In the background, myxoid stroma and edema are noted. Occasional cells showed prominent nucleoli, binucleation, and invasion into the intertrabecular spaces. Individual cell tumor necrosis and fibrin deposition were seen. Host bone entrapment was also noted consistent with low-grade chondrosarcoma WHO Grade I [Figure 3].

- Histomorphology images showing (a) a tumor comprising of well-defined lobules of hyaline cartilage along with trabecular and lamellar bone (H and E, ×200), (b) host bone entrapment (arrows) with bone destruction (H and E, ×100) and (c) the tumor shows bland looking chondrocytes with focal cellular atypia, hypercellularity, occasional prominent nucleoli (arrow), binucleation and invasion into the intertrabecular spaces with myxoid stroma and edema in the background (H and E, ×400)
Chordomas arise from the remnants of notochord which can also present in parasellar region, but they are midline and usually lack calcification. These tumors should be differentiate from chondrosarcoma as they possess remarkable difference in treatment and prognosis.
Chordomas are characterized by lobulated architecture, abundant myxoid matrix, and low cellularity. The tumor cells are large, polygonal and contain abundant eosinophilic to amphophilic cytoplasm with or without round clear vacuoles known as physaliphorous cells arranged in cords, strands, and cohesive clusters. In difficult scenario, chordoma can be differentiated from chondrosarcoma by immunohistochemical positivity for cytokeratin, epithelial membrane antigen, vimentin, S-100, and brachyury.[6]
Although surgical removal is mainstay of treatment, complexity of its location is the biggest hurdle challenging their complete resection. Based on location and direction of tumor growth, chondrosarcomas are further classified as parasellar, straddle, and posterior cranial fossa for the selection of suitable surgical approach. Parasellar variants commonly involve cranial nerves III and VI. Two of the patients in this series had cranial nerve III and VI palsies. In contrast to commonly used transnasal and transsphenoidal approach for pituitary adenomas, parasellar chondrosarcomas are resected through frontotemporal or subtemporal-infratemporal approach. Radiotherapy is questionable, but some studies showed a promising result in proton beam radiation following maximal surgical resection.[7]
To conclude, parasellar chondrosarcomas are rare skull base tumors which can be accurately diagnosed by pathologists. Host bone entrapment by atypical chondrocytes is one of the important histological features which favor the diagnosis.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- Differential diagnosis of sellar masses. Endocrinol Metab Clin North Am. 1999;28:81-117, vi.
- [Google Scholar]
- Pituitary Chondrosarcoma presenting as a sellar and suprasellar mass with parasellar extension: An unusual presentation. Iran J Pathol. 2016;11:161-6.
- [Google Scholar]
- A systematic review of intracranial chondrosarcoma and survival. J Clin Neurosci. 2009;16:1547-51.
- [Google Scholar]
- Prognostic value of MIB-1, p53, epidermal growth factor receptor, and INI1 in childhood chordomas. Neuro Oncol. 2014;16:372-81.
- [Google Scholar]
- A systematic review of proton therapy in the treatment of chondrosarcoma of the skull base. Neurosurg Rev. 2010;33:155-65.
- [Google Scholar]




