
Translate this page into:
Lhermitte-Duclos disease: A series of six cases
*Corresponding author: Asha Shenoy, Department of Pathology, Seth Gordhandas Sunderdas Medical College and King Edward Memorial Hospital, Mumbai, Maharashtra, India. shenoyasha@yahoo.co.in
-
Received: ,
Accepted: ,
How to cite this article: Kolhe AA, Shenoy A, Tayal S, Goel NA. Lhermitte-Duclos disease: A series of six cases. J Neurosci Rural Pract 2023;14:127-31.
Abstract
The Lhermitte-Duclos disease (LDD), also known as dysplastic cerebellar gangliocytoma, is a rare lesion characterized by variable enlargement of cerebellar folia. The pathological basis of LDD has long been debated, as it has overlapping features of both, a neoplasm and hamartoma. Association between LDD and Cowden syndrome (CS) has been established based on the presence of phosphatase and tensin homologue germline mutation in both. We present a series of six cases of LDD: Four females and two males, aged between 16 and 38 years, presenting with headache and imbalance on walking of 1–7 months duration. Histomorphology showed thickening and vacuolation of the molecular layer, loss of Purkinje cells, and replacement of granular cell layer by large dysplastic ganglion cells. Awareness of histological features of this rare entity and a higher level of suspicion is required for the correct diagnosis, which, in turn, should prompt thorough investigations to exclude features of associated CS. LDD is a rare entity, awareness of its histological features and correlating them with radiology is essential, especially in tiny biopsies; to render the correct diagnosis. Diagnosis of LDD warrants further clinical workup and close follow-up for the associated features of CS.
Keywords
Dysplastic gangliocytoma of cerebellum
Lhermitte-Duclos disease
Cowden disease

INTRODUCTION
Lhermitte-Duclos disease (LDD) or dysplastic cerebellar gangliocytoma is a rare benign lesion characterized by unilateral or bilateral enlargement of cerebellar folia and replacement of internal granular layer by dysplastic ganglion cells, which may present as a space-occupying lesion. Due to its insidious onset, slow progression, and good prognosis, it is now classified as Grade 1 neuroglial tumor in 2016 WHO classification of CNS tumors. It was first described by J Lhermitte and P Duclos in 1920 as the tumor with combination of a congenital malformation and a neoplasm arising from ganglion cells.[1] It has been referred by a variety of synonymous descriptions, such as cerebellar granule cell hypertrophy, diffuse hypertrophy of the cerebellar cortex, and gangliomatosis of the cerebellum.[2] Some cases of LDD are seen to be associated with the autosomal dominant Cowden disease, both of which are linked to germ line mutations in the phosphatase and tensin homologue (PTEN).[3]
CASE SERIES
A total of six cases of LDD were retrieved from neuropathology records from 2005 to 2020, of a tertiary care center. Out of these, four were female and two were male, of age ranging from 16 to 38 years. All cases presented with signs and symptoms of raised intracranial tension, along with cerebellar signs. Duration of symptoms varied from 1 month to 7 months. Four cases involved right cerebellar hemisphere and two involved left cerebellar hemisphere. Two cases additionally involved vermis and compressed fourth ventricle. Radiologically, two out of the six cases were suspected to be LDD on magnetic resonance imaging (MRI) [Figure 1a]. [Table 1] shows the clinical, radiological, and histopathological findings of individual cases.
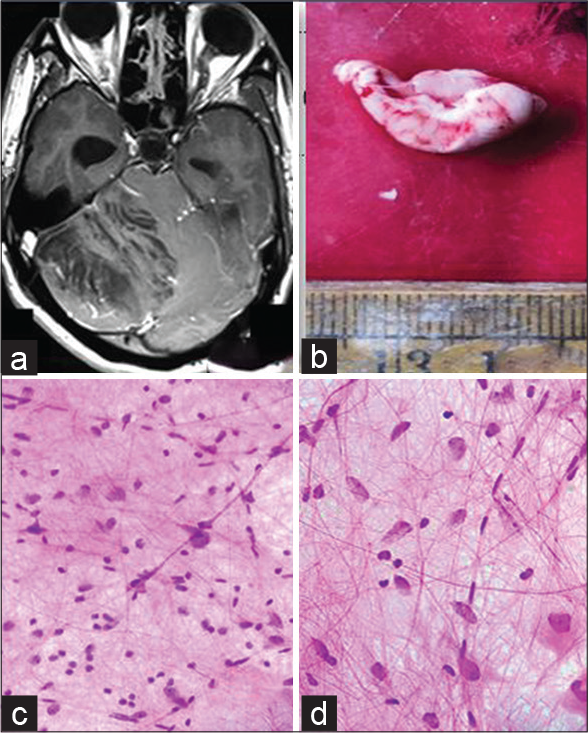
- (a) MRI-T1 weighted MRI of the brain showing a hyperintense lesion of right cerebellar hemisphere with striated folial pattern or tiger-striped appearance, (b) Gross-Enlarged cerebellar folia, (c) (squash cytology, H&E x 400) - scattered, enlarged cells with vesicular nuclei and prominent nucleoli against a fibrillary background, (d) (squash cytology, H&E x 400) - Thickened axons and capillary sized vessels.
| No. | Age/gender | Presenting complaints | Imaging | Intraoperative cytology | Gross | Microscopy | |||
|---|---|---|---|---|---|---|---|---|---|
| Site | MRI | Molecular layer | Granular layer | Others | |||||
| 1. | 16/F | Headache Vomiting |
Vermis and part of right cerebellum | Non-enhancing space-occupying lesion of mixed intensity | NA | Tissue bits resembled thickened cerebellar folia | Thickening, focal vacuolation and calcification of capillary walls | Large dysplastic neurons; scattered and in aggregates | Loss of Purkinje cells |
| 2. | 17/M | Headache Giddiness Blurring of vision |
Vermis and left cerebellum | Non-enhancing lesion compressing 4thventricle with features suggestive of LDD | NA | Occasional bit resembled cerebellar folia. | Vacuolar changes, prominent capillaries, and calcification | Replacement of granular layer by large atypical neurons forming vague nodular aggregates at places. | Recurrence after 7 years with complaints of imbalance on walking. Histopathological examination revealed similar histology. |
| 3. | 38/F | Giddiness Vertigo Headache Ataxia |
Left cerebellum | Intra-axial lesion, hypointense on T1 and hyperintense on T2 with tiger pattern suggestive of LDD | Suggestive of LDD | Multiple gray-white tissue bits | Widened and spongy change with calcification in capillary wall | Large dysplastic neurons replacing granular layer | - |
| 4. | 18/F | Headache Giddiness Ataxia |
Right cerebellum | Well-defined lesion with multiple cystic and solid components, suggestive of a glial neoplasm | Large ganglion cells with plasma cells around vessels | Cystic tissue bits with occasional bit resembling cerebellar folia | Large dysmorphic ganglion/neurons | Loss of granular cell layer | Loss of Purkinje cells, areas of gliosis and calcification. Inflammatory cells predominantly plasma cells. GFAP-+ve in gliotic fragments Synaptophysin-positive in dysplastic ganglion cells |
| 5. | 25/F | Headache Giddiness Imbalance on walking |
Right cerebellum | Space occupying lesion suggestive of Ependymoma/Glioblastoma | NA | Tissue bits resembling cerebellar folia. Cut surface is white and gritty to cut | Vacuolation and calcification | Depletion of neurons of inner granular layer, aggregates of large dysmorphic neurons. | Loss of Purkinje cells, gliosis |
| 6. | 32/M | Decreased hearing on right side Giddiness |
Right cerebellum | Mildly enhancing cerebellar mass. | Suggestive of LDD | Single Tissue bit resembling enlarged Cerebellar folia | Widened with atypical hypertrophied dysplastic ganglion cells. Vacuolation seen. | Scattered, enlarged cells with vesicular nuclei and prominent nucleoli | Thickened axons, Many capillary sized vessels seen |
Grossly, the tissue bits resembled thickened folia (five cases) [Figure 1b], whereas, in one case, they were membranous.
Squash smears of three cases, received for intraoperative assessment, showed scattered, enlarged cells with vesicular nuclei, prominent nucleoli, thickened axons, and many capillary sized vessels against a fibrillary background [Figure 1c and d].
Histopathology showed thickened and spongy molecular layer with vacuolar change and loss of granular layer and Purkinje cells [Figure 2a and b]. Aggregates and scattered large dysplastic neurons with vesicular nuclei and prominent nucleoli are shown [Figure 2c]. Prominent microvasculature and calcification were seen in two cases [Figure 2d]. A case showed prominent plasma cells amidst the clusters of dysplastic ganglion-like cells [Figure 3a and b].
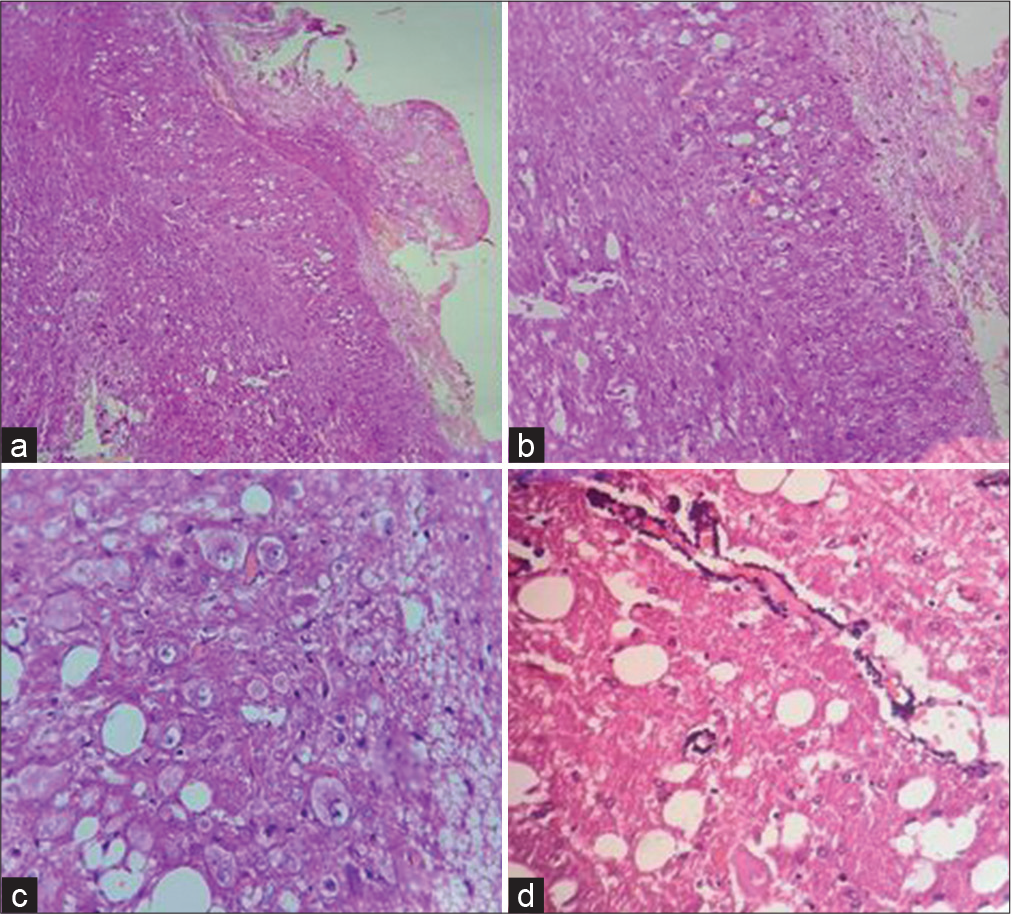
- (a) H&E x40, (b) (H&E x100) - Show enlarged cerebellar folia with vacuolation of molecular and granular layer and loss of Purkinje cells, (c) (H&E x100) - aggregates of dysplastic neurons in molecular and granular layer, (d) (H&E x400) - Shows calcified vessel wall and vacuolation in background.
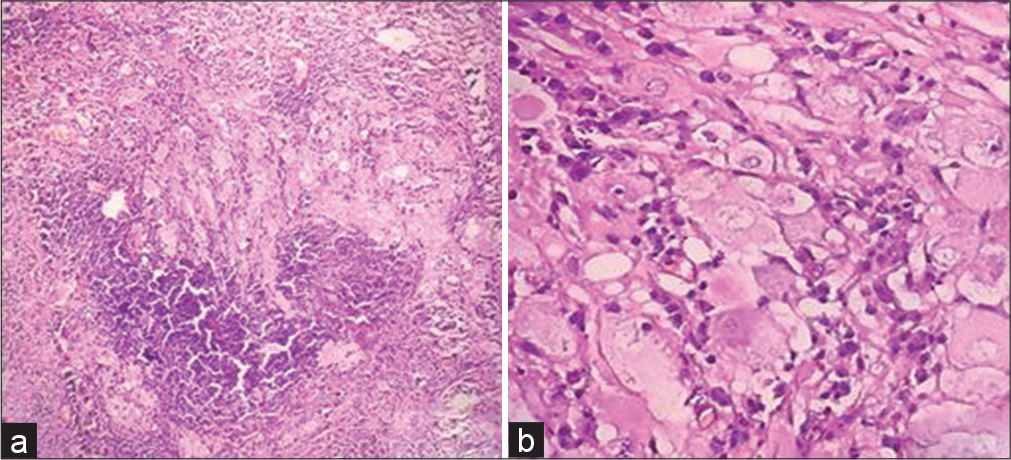
- (a) (H&E x100) - Shows aggregates of large dysmorphic neurons admixed with dense inflammation predominantly plasma cells, (b) (H&E x400) - show numerous dysplastic ganglion cells.
Clinical follow-up was unavailable except for one case (no. 2) who presented with recurrence after 7 years, at the same site, and showed similar radiological and histopathological features as before.
DISCUSSION
LDD is a rare lesion of the cerebellum of uncertain etiology and pathogenesis. Immunohistochemical studies, recurrence of the lesion following initial surgery and development of lesion in patients with previously normal MRI supports its neoplastic origin, whereas, malformative histopathological features, very low or absent mitosis, and absence of progression in some cases support its hamartomatous nature.[1,2,4]
It can be familial, but is more commonly sporadic.[3] Ambler et.al first described the familial association in 1969.[5] In 1991, Padberg et al. described its association with Cowden syndrome (CS).[6] CS is an autosomal dominant disorder which is characterized by multiple hamartomas derived from all three germ layers, intestinal polyposis, palmoplantar keratoses, oral papillomas and increased incidence of malignant tumors, most commonly breast, thyroid, genitourinary, and endometrial tumors, due to abnormalities in 10q23. Few studies have suggested, adult-onset LDD as the pathognomonic feature of CS, though isolated (non-syndromic) cases are also reported.[3] LDD, in most cases, is related to loss of the inhibitory influence of PTEN on the PI3K pathway, resulting in deleterious effects on neuronal migration and regulation of cell size; thus, best considered a hypertrophic phenomenon superimposed on a developmental malformation.[7] These PTEN mutations are found to be more commonly associated with the late-/adult-onset LDD as compared to the childhood-onset disease.[3,7] Germline mutations in the PTEN gene on chromosome 10 were found to be linked to both conditions (CD and LDD) and the loss of inhibitory influence of PTEN on the PI3K pathway and activation of the AKT/mTOR signaling were suggested to be the responsible pathogenetic mechanism.[8] Although these associations are well documented, the precise relationships between LDD, CD, and germline PTEN mutations are less clear, as not all patients with LDD develop the clinical manifestations of CD, and some patients with LDD lack germ line mutations in PTEN.[7] Furthermore, not all patients with germline PTEN mutations manifest CD.[7] In our study, there were no findings associated with CS at the time of presentation and later patients were lost to follow-up.
LDD has been reported in all age groups (3 years–74 years). Most cases of LDD occur in adults, usually in the third and fourth decades. In our series, the youngest age of presentation was 16 years and oldest was 38 years. No gender predilection was observed in the literature; however, our study showed female preponderance.[1,2,9]
Clinical manifestations are related to mass effect and secondary to obstructive hydrocephalus. Dysmetria and ataxia are common.[9] Most of our cases presented with headache and ataxia. Pre-operative symptoms can be of variable duration.
On MRI, LDD shows pathognomonic “tiger-striped” appearance of involved cerebellum due to close apposition of thickened cerebellar folia, which has lost secondary arborization, thus, explaining the alternation of different tissue intensities.[10] Two cases in our study showed these features, whereas others were diagnosed as a glial tumor on MRI. Some studies have reported a false “tiger pattern” sign in a medulloblastoma, wherein it was misdiagnosed as LDD. Therefore, to improve the diagnostic accuracy, functional imaging other than conventional MRI is necessary. Although LDD is a tumor with a relatively low malignant potential (WHO I), it can still cause high perfusion on perfusion-weighted imaging. In addition to the local hyperperfusion caused by the blood vessels around the lesion, the tumor itself could invade the blood–brain barrier, resulting in increased overall tumor perfusion which is also a feature of other cerebellar tumors, including pilocytic astrocytoma and medulloblastomas. However, the use of perfusion-weighted imaging for diagnosing LDD requires additional confirmation. On magnetic resonance spectroscopy, the cholinesterase/noradrenaline (Cho/NAA) ratio is usually used for judging the degree of malignancy in brain tumors. An increase in the Cho peak generally indicates that the cell membrane is renewed quickly and the cell density is high. A decrease in NAA usually reflects neuronal loss. In malignant brain tumors, such as high-grade gliomas, the Cho peak is significantly high, the NAA peak is significantly low, and the Cho/NAA ratio is high. In LDD patients, the Cho/NAA ratio is usually low. The appearance of the Lac peak, low Cho/NAA ratio, and comprehensive presence of the conventional MRI tiger striped sign can improve the diagnostic accuracy of LDD.[11]
The MRI features often correlate with the histopathological features of LDD. The central core of T1 hypointensity and T2 hyperintensity corresponds to the thinned white matter, widened granular cell layer, and the inner portions of the dysplastic molecular layer. The outer thinner layer on MR images (T1 isointense and T2 iso- to hypointense) can be attributed to the outer molecular layer and the leptomeninges which are associated with abnormal vessels and areas of calcification.[12,13]
In spite of these developments in the imaging modalities which have made pre-operative diagnosis of LDD easier, histopathology is still required for definitive diagnosis. Grossly, it is pale ill-circumscribed lesion and shows enlargement of cerebellar cortex on cut section.[2] Microscopy shows enlargement of molecular and internal granular layer with relative preservation of cerebellar foliar architecture.[2] Molecular layer is broadened by numerous, enlarged, and irregularly myelinated axons, often arranged in parallel arrays and arise from the abnormal hypertrophied neurons comprising the internal granular layer. In addition, absence of Purkinje cells, calcification of vessel wall, and vacuolar changes of molecular layer are sometimes observed.[2,9] Immunophenotypically, dysplastic neuronal cells are positive for synaptophysin.[2,4] Loss of PTEN protein expression is seen.[2,3,7]
Differential diagnosis of LDD includes gangliocytomas (WHO Grade1) which are composed of ganglion cells. However, frequent location of these tumors in the temporal lobe, presentation with epilepsy, and a solid cystic lesion on imaging help to differentiate the two entities.[14]
Radiological and histopathological features of this disease are distinctive and very rarely confused with other tumors, but as the lesion is very rare a high degree of suspicion is required for diagnosing LDD.
The treatment is usually radical surgical excision of the lesion. Total excision is difficult due to progressive transition and ill-defined margins between normal and pathological tissue. The location and volume of sacrificed cerebellar tissue are associated with loss of function. Small lesions in the vermis may cause no symptoms or deficit, but larger lesions of midline structures, such as the uvula, nodule, and flocculus, involving vestibular fibers in the cerebellum may lead to clumsiness or problems with coordination of movement, with truncal ataxia or staggering gait. Gaze-evoked or positional nystagmus occurs while the lesion damages the vestibular projections from the brainstem to the flocculonodular lobe. Cerebellar mutism, a transient complication with an unclear anatomical substrate, usually happens after the removal of midline tumors involving the vermis. Hence, subtotal and partial resection of LDD may be acceptable due to its remarkably slow-growing nature.[15] Radiation therapy has been reported as non-effective in avoiding the growth of the lesion.[2,9]
Prognosis is favorable and mortality is low, though recurrences may occur due to subtotal resection.
CONCLUSION
It is extremely important for the pathologist to be aware of the variable clinical and histopathological presentations of this rare disease, particularly to differentiate it from the low-grade glial and glioneuronal tumors. Its diagnosis warrants the screening for associated CS.
Declaration of patient consent
Patient’s consent not required as there are no patients in this study.
Conflicts of interest
There are no conflicts of interest.
Financial support and sponsorship
Nil.
References
- Lhermitte-duclos disease (dysplastic cerebellar gangliocytoma): A malformation, hamartoma or neoplasm? Acta Neurol Scand. 2002;105:137-45.
- [CrossRef] [PubMed] [Google Scholar]
- Dysplastic cerebellar gangliocytoma (lhermitteduclos disease) In: Louis DN, ed. WHO Classification of Tumors of the Central Nervous system (4th ed). Lyon: IARC; 2016. p. :142-3.
- [Google Scholar]
- Germline inactivation of PTEN and dysregulation of the phosphoinositol-3-kinase/akt pathway cause human Lhermitte-duclos disease in adults. Am J Hum Genet. 2003;73:1191-8.
- [CrossRef] [PubMed] [Google Scholar]
- Immunohistochemistry and proliferative activity in Lhermitteduclos disease. Acta Neuropathol. 1992;84:570-3.
- [CrossRef] [PubMed] [Google Scholar]
- Lhermitte-duclos disease (granule cell hypertrophy of the cerebellum) pathological analysis of the first familial cases. J Neuropathol Exp Neurol. 1969;28:622-47.
- [CrossRef] [PubMed] [Google Scholar]
- Lhermitte-duclos disease and Cowden disease: A single phakomatosis. Ann Neurol. 1991;29:517-23.
- [CrossRef] [PubMed] [Google Scholar]
- Lhermitte-duclos disease: A report of 31 cases with immunohistochemical analysis of the PTEN/AKT/mTOR pathway. J Neuropathol Exp Neurol. 2005;64:341-9.
- [CrossRef] [PubMed] [Google Scholar]
- Lhermitte-duclos disease: A rare lesion with variable presentations and obscure. Histopathology Turk Patoloji Derg. 2018;34:92-9.
- [Google Scholar]
- Lhermitte-duclos disease and Cowden disease: Clinical and genetic study in five patients with LhermitteDuclos disease and literature review. Acta Neurochir (Wien). 2004;146:679-90.
- [CrossRef] [PubMed] [Google Scholar]
- Cowden disease and Lhermitte-duclos disease: An update. Case report and review of the literature. Neurosurg Focus. 2006;20:E6.
- [CrossRef] [PubMed] [Google Scholar]
- MR imaging features of Lhermitte-duclos disease. Case reports and literature review. Medicine (Baltimore). 2022;101:e28667.
- [CrossRef] [PubMed] [Google Scholar]
- Lhermitte-duclos disease: A case report with radiologic-pathologic correlation. Radiol Case Rep. 2019;14:734-9.
- [CrossRef] [PubMed] [Google Scholar]
- Dysplastic cerebellar gangliocytoma: A description of two cases. Quant Imaging Med Surg. 2021;11:4695-9.
- [CrossRef] [PubMed] [Google Scholar]
- Lhermitte-duclos disease: A rare entity. Med J Armed Forces India. 2016;72:147-9.
- [CrossRef] [PubMed] [Google Scholar]
- The surgical resection of dysplastic cerebellar gangliocytoma assisted by intraoperative sonography: Illustrative case. J Neurosurg Case Lessons. 2021;2:CASE21451.
- [CrossRef] [Google Scholar]




