
Translate this page into:
Involvement of the Spinal Cord in Mitochondrial Disorders
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
This review aims at summarising and discussing the current status concerning the clinical presentation, pathogenesis, diagnosis, and treatment of spinal cord affection in mitochondrial disorders (MIDs). A literature search using the database Pubmed was carried out by application of appropriate search terms and their combinations. Involvement of the spinal cord in MIDs is more frequent than anticipated. It occurs in specific and non-specific MIDs. Among the specific MIDs it has been most frequently described in LBSL, LS, MERRF, KSS, IOSCA, MIRAS, and PCH and only rarely in MELAS, CPEO, and LHON. Clinically, spinal cord involvement manifests as monoparesis, paraparesis, quadruparesis, sensory disturbances, hypotonia, spasticity, urinary or defecation dysfunction, spinal column deformities, or as transverse syndrome. Diagnosing spinal cord involvement in MIDs requires a thoroughly taken history, clinical exam, and imaging studies. Additionally, transcranial magnetic stimulation, somato-sensory-evoked potentials, and cerebro-spinal fluid can be supportive. Treatment is generally not at variance compared to the underlying MID but occasionally surgical stabilisation of the spinal column may be necessary. It is concluded that spinal cord involvement in MIDs is more frequent than anticipated but may be missed if cerebral manifestations prevail. Spinal cord involvement in MIDs may strongly determine the mobility of these patients.
Keywords
Mitochondrial
mtDNA
myelon
myelopathy
respiratory chain
spinal cord
transverse syndrome

INTRODUCTION
Mitochondrial disorders (MIDs) are usually multisystem disorders, either at onset or evolve from a mono-organ disease to a mitochondrial multi-organ disorder syndrome (MIMODS) as the disease progresses.[1] The organs most frequently affected in MIMODS are the skeletal muscle, the central nervous system (CNS), the peripheral nerves, the eyes, ears, endocrine organs, the heart, lungs, intestines, kidneys, blood, bone, and the skin.[2] Concerning the CNS involvement, affection of the brain dominates research interests. However, CNS involvement is not restricted to the brain but can be found also in the spinal cord. Spinal cord involvement in MIDs most frequently occurs together with affection of the brain but may occur without cerebral abnormalities as well. Considering and recognizing spinal cord involvement in MIDs is essential since it may influence the management of these patients. This review aims at summarizing and discussing the current status concerning the clinical presentation, pathogenesis, diagnosis, and treatment of spinal cord affection in MIDs.
MATERIALS AND METHODS
Data for this systematic review were identified by searches of MEDLINE for references of relevant articles. Search terms used were all acronyms known for specific MIDs (n = 50) and the terms “MID,” “mtDNA,” “encephalomyopathy,” and “mitochondrion” in individual combination with the terms “spinal cord,” “myelon,” “spine,” “myelopathy,” “scoliosis,” “transverse syndrome,” or “spinal colum,” results of the searches were screened for potentially relevant studies by application of inclusion and exclusion criteria for the full texts of relevant studies. Included were only original articles about humans and published between 1966 and 2017. Only randomized controlled trials, observational studies with controls, case series, and case reports were included. Reviews, editorials, and letters were excluded. In addition, reference lists of retrieved studies were checked for reports of studies not detected on the electronic search. Websites checked for additional information with regard to spinal cord involvement in MIDs were MITOMAP (https://www.mitomap.org/foswiki/bin/view/MITOMAP/ClinicalPhenotypesRNA), Neuromuscular Disease Center Database (http://neuromuscular.wustl.edu/mitosyn.html#merrf), and MitoTools (http://www. mitotool.org/database.html).
RESULTS
Classification
Spinal cord involvement in a MID is in the majority of the cases part of a multisystem disease involving not only the spinal cord but also the cerebrum and organs other than the CNS. Only in rare cases, the spinal cord may be exclusively affected, which is the case in Leigh syndrome (LS)[3] and some patients mimicking motor neuron disease.[45] Spinal cord involvement may be also classified according to the affected structure within the myelon. Spinal cord affection may involve only motor fibers, only sensory fibers, only autonomic fibers, or other fibers. Abnormalities on imaging studies can be differentiated from histological abnormalities.
Clinical presentation
Spinal cord involvement in MIDs may manifest as monoparesis, paraparesis, quadruparesis, sensory disturbances in a segmental distribution, hypotonia, spasticity, as urinary or defecation dysfunction, spinal column deformities (increased kyphosis, increased lordosis, and scoliosis), or as sensory or motor transverse syndrome.
Specific mitochondrial multi-organ disorder syndrome
Mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes syndrome
In a retrospective series of 69 mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) patients, six patients showed neuronal loss in anterior and posterior horns and degeneration of corticospinal tracts and the posterior or lateral columns.[6] In a 24-year-old female with MELAS/myoclonic epilepsy with ragged-red fibers (MERRF) overlap syndrome postmortem investigations of the spinal cord showed gliosis of the posterior horns.[7]
Myoclonic epilepsy with ragged-red fibers syndrome
In a 16-year-old Mexican girl with MERRF syndrome due to the m. 8344A>G variant postmortem autopsy studies revealed neuronal loss in Clarke's nucleus and the posterior horns of the cervical and thoracic spinal cord. The spinal cord's white matter displayed normal myelination including the spinocerebellar tracts and dorsal columns.[8] In an 18-year-old male with MERRF syndrome, due to the m. 8344A>G variant, autopsy studies after death from pneumonia showed neuronal loss and gliosis of the spinal cord.[9] In a 16-year-old female and a 21-year-male with MERRF syndrome autopsy revealed mild neuronal loss in the anterior and posterior horns but severe neuronal loss in Clarke's column.[10] There was also severe degeneration in the gracile fasciculus and posterior spinocerebellar tract, moderate degeneration in the cuneate fasciculus, and mild degeneration in the lateral and anterior corticospinal, and anterior spinocerebellar tracts.[10] In a 45-year-old female with MERRF/LS overlap syndrome and neuropathy due to the m. 8344A>G variant, magnetic resonance imaging (MRI) of the spinal cord showed severe atrophy of the myelon.[11] In a Chinese patient with MERRF syndrome due to the variant m. 8344A>G, postmortem histological studies revealed a heteroplasmy rate of 80% in the anterior horn cells.[12] In an autopsy study of a patient with MERRF syndrome due to the pathogenic variant m.8344A>G application of antibodies against subunits of Complex-III and Complex-IV of the respiratory chain revealed decreased expression of subunit-2 of Complex-IV.[13]
Kearns–Sayre syndrome
In two patients with Kearns–Sayre syndrome (KSS) due to an mtDNA deletion, respectively, duplication and pathological features as in LS were found in the spinal cord.[14] In Patient A (deletion) there was mild microvacuolation and gliosis of the dorsal columns in the cervical segment.[14] In Patient B (tandem duplication), the corticospinal tracts were symmetrically pale with marked microvacuolation.[14] There was also mild microvacuolation and myelin pallor in the dorsal columns in all segments.[14] In addition, dorsal columns contained focal areas of capillary hyperplasia.[14] These capillaries were lined by swollen endothelial cells and separated by damaged neutrophils.[14] There was some infiltration of macrophages and reactive astrocytosis, resembling pathological features as described in LS.[14] Postmortem, neuropathological studies in a 22-year-old male with KSS revealed mild spongiosis and astrocytosis of the long descending tracts.[15] Dorsal columns of the cervical cord showed loss of myelin and axonal loss.[15] In another KSS case, autopsy disclosed severe demyelination of the initial few millimeters of the spinal anterior roots distal to the glial-Schwann cell junction.[16] Mild demyelination was observed in the dorsal spinal roots.[16]
Leigh syndrome
LS is an early-onset progressive MID, characterized by elevated lactate and pyruvate and bilateral symmetric hyperintense lesions in the basal ganglia, thalamus, brainstem, cerebral white matter, or spinal cord on T2-weighted MRI.[17] LS is the most frequent disorder of energy production in children.[18] Typical abnormalities on spinal MRI include bilaterally symmetric, high-signal alterations in the spinal cord.[1920] Rarely, the typical symmetric cerebral symmetric lesions in LS are confined to the spinal cord and are absent in the brain.[3] In a 6-month-old Hispanic male with early-onset LS, involvement of the cervical spinal cord and brainstem were responsible for the initial clinical presentation.[21] At age 2 months, the cerebral MRI was normal. Repeat MRI of the cerebrum and the cervical spine at age 3.5 months showed abnormal signal intensity and restricted diffusion involving the brainstem (pons and medulla) and upper cervical spinal cord.[21] In a 3-year-old female with the clinical diagnosis of LS who died suddenly and unexplained, autopsy showed symmetrical, necrotizing lesions scattered within the spinal cord's gray matter.[22] In a series of 5 children with LS, isolated affection of the spinal cord without brain abnormalities was described in a 17-month-old female at autopsy.[3] Altogether, 4 of the 5 patients had spinal cord involvement [Figure 1].[3] T2-hyperintensities extending from the medulla to the cervical spinal cord were also found in a 15-month-old female with LS.[23] A 10-year-old patient with LS and intractable Lennox–Gastaut syndrome had mild demyelination, proliferation of capillaries, and gliosis of the entire spinal cord at autopsy.[24] Postmortem histopathological investigations in an 8-year-old male with LS revealed symmetrical microcyst formation, vascular proliferation, proliferation of astrocytes, fibrillary gliosis, and partial destruction of the myelin sheaths with relative preservation of the neuronal cell bodies in the spinal cord.[25]

- T2-weighted sagittal magnetic resonance imaging (repetition time 3000 ms/echo time 120 ms) depicting increased signal change in cervical and thoracic cord in a patient with Leigh syndrome[3]
Leukoencephalopathy, brain stem and spinal cord involvement and lactic acidosis
Leukoencephalopathy, brain stem and spinal cord involvement and lactic acidosis (LBSL) is a rare specific MID due to mutations in the DARS2 gene encoding mitochondrial aspartyl-tRNA synthetase.[26] Clinically, patients present with childhood or juvenile-onset intellectual decline (learning dysfunction), slowly progressive ataxia, pyramidal signs (spasticity), and dorsal column dysfunction.[27] MRI of the spine shows inhomogeneous T2-hyperintensities in the spinal tract.[28] Proton-magnetic resonance spectroscopy (MRS) of the frontal and cerebellar white matter shows elevated lactate, reduced N-acetyl-aspartate, increased myoinositol, and mildly elevated choline.[29] Neuropathological investigations typically show vacuolar changes in the white matter of the cerebrum and spinal cord.[30] MRI does not necessarily reflect clinical presentation and the histological findings why it should not be used to assess disease severity or prognosis.[31] In a 1-year-old male with psychomotor delay, quadruplegia, deafness, and anemia due to a mutation in the DARS2 gene, spinal MRI revealed calcifications of the spinal cord.[32] A 35-year-old female with mild balance impairment since her early teenage years, developed slowly progressive unsteady gait with lower limb weakness and stiffness since age 20 years.[33] Neurological examination revealed spastic paraparesis with marked impairment of vibration and position perceptions and waddling gait.[33] Spinal MRI in this patient showed T2-hyperintensities in the posterior columns, anterior and posterior spinocerebellar tracts, and lateral corticospinal tracts [Figure 2], indicating continuous involvement of the corresponding pathways in the brainstem.[33]

- T2-weighted magnetic resonance imaging of the spine in a leukoencephalopathy, brain stem and spinal cord involvement and lactic acidosis patient showing hyperintensities of the dorsal column (sagittal section) and hypointensities of the anterior and posterior spinocerebellar tracts (black arrow and black arrowhead), nuclei and fasciculi gracilis (white arrowhead), and the pyramidal decussation (white arrow) [reproduced with permission from Lan et al., J Neurol Sci 2017])[33]
Pontocerebellar hypoplasia
Histological examination of the spinal cord of an infant with pontocerebellar hypoplasia (PCH) due to a mutation in the SLC25A46 gene who had died 1 day after birth revealed loss of motor neurons in the anterior horns.[34] The remaining motor neurons had a “ballooning” appearance.[34] The sister of this patient also had PCH, died at age 18 days, and had mild reduction of motor neurons in the anterior horns and “ballooning” in some of the remaining motor neurons.[34] Knock-down of the SLC25A46 gene in zebrafish also caused spinal motor neuron loss.[35]
Infantile-onset spinocerebellar ataxia
Infantile-onset spinocerebellar ataxia (IOSCA) is a severe autosomal recessively inherited neurodegenerative disorder characterized by progressive atrophy of the cerebellum, brainstem and spinal cord, and sensory axonal neuropathy.[36] IOSCA is due to mutations in the nuclear gene C10orf2 encoding the twinkle protein.[36] In two autopsy cases, CNS atrophy was the dominant morphological feature, which was most severe in the spinal cord.[37] Atrophy there was most prominent near the dorsal roots, the dorsal columns, and the posterior spinocerebellar tracts.[37] There was also severe loss of neurons in the dorsal nucleus (Clarke's column).[37] In a study of 17 IOSCA patients, 9 underwent MRI of the spinal cord of which 4 showed moderate atrophy of the upper cervical cord.[38]
Mitochondrial recessive ataxia syndrome
Mitochondrial recessive ataxia syndrome (MIRAS) is a specific MID, clinically characterized by adult-onset ataxia, neuropathy, psychiatric abnormalities, and cognitive impairment. MIRAS is most frequently due to mutations in POLG1. In a 56-year-old female with MIRAS, who deceased from pulmonary embolism, neuropathological workup revealed atrophy of the posterior columns of spinal cord, particularly of the gracile tract and the posterior spinocerebellar tracts [Figure 3].[39] Clarke's dorsal nuclei showed some neuronal loss, while anterior horns or intermediolateral columns were spared.[39]
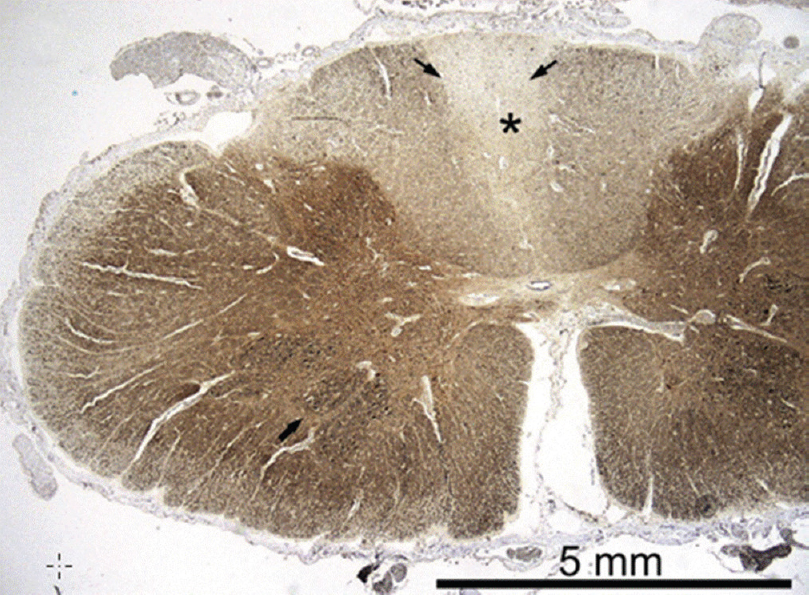
- Autopsy sample of the spinal cord at level C7; neurofilament immunohistochemistry shows a clear pallor of the posterior columns (asterisk), and also the posterior spinocerebellar tracts are slightly fiber-depleted (thin arrow). The motor neurons in the anterior horn are preserved (thick arrow) Paraffin sections, SMI-311 IHC ×10. [reproduced with permission from Palin et al., J Neurol Sci 2012][39]
Leber's hereditary optic neuropathy
In a study of 46 patients with leber's hereditary optic neuropathy (LHON), thoracic kyphosis was found in 7 of them.[40] In a single patient with LHON, due to the variant m.14484T>C in the ND6 gene, spinal MRI showed diffuse demyelinating lesions extending from T6 to T11.[41] In a patient with LHON spinal cord lesions due to concomitant neuromyelitis optica (NMO) have been reported.[42] In patients with LHON and features of multiple sclerosis (MS), respectively NMO, it is conceivable that “demyelinating” lesions are in fact stroke-like lesions attributable to the MID with some clinical and instrumental features suggesting MS, respectively, NMO. In a patient with LHON and MS, new “inflammatory lesions” were seen on MRI of the brain and spinal cord 14 months after discontinuation of natalizumab.[43] Particularly, in seronegative “NMO” patients, spinal cord involvement in LHON should be suspected.
Chronic progressive external ophthalmoplegia
In a 55-year-old female with a paternal history of ptosis, chronic progressive external ophthalmoplegia (CPEO), and ptosis developed since age 40 years.[44] These clinical manifestations were due to a mutation in the C10orf2 gene.[44] By age 53 years, she experienced sudden-onset ascending paresthesias from lower limbs to mid trunk without involvement of the sphincters.[44] On neurologic examination, she exhibited a Lhermitte's sign, 3/5 MRC shoulder abduction and elbow flexion weakness, suspended loss of touch and temperature at C7–C8 levels, impaired proprioception in toes, and lower limb brisk reflexes. Spinal MRI, performed 4 weeks after clinical onset, showed a gadolinium-unenhanced T2-hyperintensity along the posterior columns extending bilaterally for 13 mm at C7–T1 levels.[44]
Nonspecific mitochondrial multi-organ disorder syndrome
In a study of 33 patients with a genetically confirmed MID (mtDNA point mutations or deletions [n = 21], SURF1 mutations [n = 7], POLG1 mutations [n = 5]), 4 of 6 patients undergoing MRI of the spinal cord had nonspecific signal changes.[45] In a study of six patients carrying ISCA2 mutations, one had signal abnormalities in the cervical spinal cord.[46] In a single patient with MIMODS due to a tRNA (Glu) mutation causing Complex-I deficiency, neuropathological investigations at autopsy revealed descending anterograde degeneration in the medullary pyramids and corticospinal tracts.[47] In a 66-year-old female carrying a POLG1 mutation, neuropathological studies at autopsy revealed profound dorsal column and dorsal spinocerebellar tract degeneration.[48] Autopsy after death at age 59 years from pneumonia in a patient carrying a POLG1 mutation revealed myelin loss of the gracile fascicle in the cervical spinal cord.[49] Pure motor dysfunction manifesting with quadriparesis and involvement of the cranial nerves has been repeatedly reported in MIDs, which may mimic motor neuron disease.[45] This mimicry may be due to involvement of the anterior horn cells in the mitochondrial defect.
Diagnosis
Diagnosis of spinal cord involvement in MIDs requires an extensive individual history, a thorough clinical examination, imaging investigations, cerebrospinal fluid (CSF) investigations, transcranial magnetic stimulation (TMS), somatosensory-evoked potentials (SSEPs), and neuropathological work work-up in case of decease. MRI may show atrophy, T2-hypo- or hyperintensities, enhancing (inflammatory) lesions, restricted diffusion, demyelination, or calcifications. MRS may show a lactate peak. CSF investigations may be normal, may show lactic acidosis, or may reveal mild or marked pleocytosis, IgG production, and positive oligoclonal bands.[50] TMS may show prolongation of the central conduction time and of MEP latencies. Affection of the spinal cord may be documented by delayed SSEPs.[51] Neuropathological investigations may show atrophy, neuronal loss, gliosis, inflammation, edema, cyst formation, or vacuolisation. Blood tests may show lactic acidosis, creatine-kinase elevation, or elevation of pyruvate.
Treatment
Treatment of spinal cord involvement in MIDs is usually supportive, including physical therapy and rehabilitation to improve motor function, and occasionally there might be a need for special education or speech therapy.[52] Rehabilitation and physical therapy are helpful in the prevention of secondary complications such as contractures and scoliosis.[52] In case of spinal deformities surgical corrections may be an option but this procedure has been carried out only in single cases. In a patient with LS posterior spinal fusion as a palliative procedure resulted in activation of the underlying MID and finally deterioration of the phenotype.[53] In case of urinary retention, placement of a transurethral catheter or implantation of a suprapubic catheter may be necessary.
DISCUSSION AND CONCLUSIONS
This review shows that the spinal cord is involved in various MIDs. Spinal cord involvement is most frequently reported in LBSL, LS, MERRF, KSS, IOSCA, MIRAS, PCH, and MIMODS, and more rarely in MELAS, CPEO, and LHON. Clinical manifestations of spinal cord involvement include weakness, sensory and autonomic disturbances, sphincter dysfunction, or transverse syndrome. These abnormalities may overlap with cerebral manifestations in case of cerebral involvement. In some cases, spinal cord affection may be the initial manifestation of a MID, as in LS.[21] Some cases may present with features of a motor neuron disease, thus mimicking spinal muscular atrophy or amyotrophic lateral sclerosis.[45] Rarely, spinal cord involvement may manifest as sensory or motor transverse syndrome. With progression of the MID, spinal cord involvement may advance as well.
There is no uniform pattern of spinal cord involvement in MIDs, but most frequently the dorsal columns, the corticospinal tracts, or the spinocerebellar tracts are involved. Spinal cord involvement in MELAS is characterized by degeneration and neuronal loss of anterior and posterior corticospinal tracts. Spinal cord involvement in MERRF is characterized by neuronal loss in Clarke's column and the anterior and posterior horn and by gliosis of the corticospinal tracts, the spinocerebellar tracts, and the dorsal columns. Spinal cord involvement in KSS is characterized by vacuolation and gliosis of the dorsal columns and the corticospinal tracts.[14] Affection of the spinal cord in LS may manifest on imaging as bilaterally symmetric T2-hyperintensities of the cord and histologically as symmetrical microcyst formation, vascular proliferation, proliferation of astrocytes, fibrillary gliosis, and destruction of the myelin sheaths.[25] There may be necrosis of the gray matter, demyelination, capillary proliferation, and gliosis of the cervical cord.[24] Spinal cord lesions in LBSL on imaging include T2-hyperintensities in the posterior columns, in the anterior and posterior spinocerebellar tracts, and the lateral corticospinal tract. In single cases, calcifications may be seen. Histologically, vacuolation can be found. Spinal cord involvement in PCH includes loss of anterior horn cells and ballooning of neurons.[34] The spinal cord in IOSCA is atrophic, particularly near the dorsal roots, the dorsal columns, and the posterior spinocerebellar tracts.[37] Spinal cord abnormalities in MIRAS include atrophy of posterior columns and the spinocerebellar tracts and neuronal loss in Clarke's dorsal nuclei.[39]
Diagnosing spinal cord involvement in MIDs requires the application of spinal MRI, spinal computed tomography, spinal MRS, SSEPs, TMS, CSF investigations, and autopsy. Treatment of spinal cord involvement relies on the same measures as applied for MIDs in general.[54] Transverse syndromes may be managed as transverse syndromes due to other causes. In case of secondary spinal column abnormalities, orthopedic measures may be helpful. For the clinician, it is important to consider spinal cord involvement in MIDs and to initiate appropriate diagnostic steps if the clinical presentation suggests spinal cord involvement and if cerebral lesions are absent or do not explain the clinical presentation. Spinal cord involvement in MIDs must be considered as a differential diagnosis of motor neuron disease, transverse syndrome, MS, neuromyelitis optica, myelitis, ADEM, or myelopathy. Imaging modalities should not be applied to assess severity or prognosis of spinal cord lesions in MIDs.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- Genetic counselling for maternally inherited mitochondrial disorders. Mol Diagn Ther. 2017;21:419-29.
- [Google Scholar]
- Multisystem disease, including eosinophilia and progressive hyper-creatine-kinase-emia over 10 years, suggests mitochondrial disorder. Case Rep Neurol. 2017;9:69-75.
- [Google Scholar]
- Atypical presentations of Leigh syndrome: A case series and review. Pediatr Neurol. 2005;32:334-40.
- [Google Scholar]
- Mitochondrial disorders may mimic amyotrophic lateral sclerosis at onset. Sultan Qaboos Univ Med J. 2016;16:e92-5.
- [Google Scholar]
- Mitochondriopathy as a differential diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2002;3:219-24.
- [Google Scholar]
- Melas: An original case and clinical criteria for diagnosis. Neuromuscul Disord. 1992;2:125-35.
- [Google Scholar]
- AMELAS/MERRF phenotype associated with the mitochondrial DNA 5521G>A mutation. J Neurol Neurosurg Psychiatry. 2010;81:471-2.
- [Google Scholar]
- Tissue distribution and disease manifestations of the tRNA(Lys) A->G(8344) mitochondrial DNA mutation in a case of myoclonus epilepsy and ragged red fibres. Acta Neuropathol. 1995;90:328-33.
- [Google Scholar]
- Neuropathology of myoclonus epilepsy associated with ragged-red fibers (Fukuhara's disease) Acta Neuropathol. 1988;75:433-40.
- [Google Scholar]
- Association of the mitochondrial 8344 MERRF mutation with maternally inherited spinocerebellar degeneration and leigh disease. Neurology. 1996;46:219-22.
- [Google Scholar]
- Myoclonic epilepsy and ragged red fibers (MERRF) syndrome: Selective vulnerability of CNS neurons does not correlate with the level of mitochondrial tRNAlys mutation in individual neuronal isolates. J Neurosci. 1997;17:7746-53.
- [Google Scholar]
- Immunohistochemical demonstration of spinal ventral horn cells involvement in a case of “myoclonus epilepsy with ragged red fibers” (MERRF) Clin Neuropathol. 2000;19:200-7.
- [Google Scholar]
- Kearns-sayre syndrome associated with mitochondrial DNA deletion or duplication: A molecular genetic and pathological study. J Neurol Sci. 1995;131:78-87.
- [Google Scholar]
- Demyelinating radiculopathy in the Kearns-Sayre syndrome: A clinicopathological study. Ann Neurol. 1980;8:373-80.
- [Google Scholar]
- Homozygous p.V116* mutation in C12orf65 results in leigh syndrome. J Neurol Neurosurg Psychiatry. 2016;87:212-6.
- [Google Scholar]
- Widening the heterogeneity of leigh syndrome: Clinical, biochemical, and neuroradiologic features in a patient harboring a NDUFA10 mutation. JIMD Rep. 2017;37:37-43.
- [Google Scholar]
- White matter involvement in mitochondrial diseases. Mol Genet Metab. 2005;84:127-36.
- [Google Scholar]
- MR findings in leigh syndrome with COX deficiency and SURF-1 mutations. AJNR Am J Neuroradiol. 2002;23:1095-100.
- [Google Scholar]
- Early spinal cord and brainstem involvement in infantile Leigh syndrome possibly caused by a novel variant. J Child Neurol. 2013;28:1681-5.
- [Google Scholar]
- Sudden death in Leigh syndrome: An autopsy case. Am J Forensic Med Pathol. 2012;33:259-61.
- [Google Scholar]
- Leigh syndrome with COX deficiency and SURF1 gene mutations: MR imaging findings. AJNR Am J Neuroradiol. 2003;24:1188-91.
- [Google Scholar]
- Subacute necrotizing encephalomyelopathy (Leigh disease): Report of a case with lennox-gastaut syndrome. Brain Dev. 1985;7:500-4.
- [Google Scholar]
- Pathological findings in subacute necrotizing encephalomyelopathy (Leigh's disease) (author's transl) Zentralbl Allg Pathol. 1975;119:488-94.
- [Google Scholar]
- Mitochondrial aspartyl-tRNA synthetase deficiency causes leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation. Nat Genet. 2007;39:534-9.
- [Google Scholar]
- Leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation in the first polish patient. Brain Dev. 2011;33:713-7.
- [Google Scholar]
- Leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation is associated with cell-type-dependent splicing of mtAspRS mRNA. Biochem J. 2012;441:955-62.
- [Google Scholar]
- Leukoencephalopathy with brain stem and spinal cord involvement and high lactate: A genetically proven case with distinct MRI findings. J Neurol Sci. 2008;273:118-22.
- [Google Scholar]
- Neuropathology of leukoencephalopathy with brainstem and spinal cord involvement and high lactate caused by a homozygous mutation of DARS2. Brain Dev. 2013;35:312-6.
- [Google Scholar]
- Leukoencephalopathy with brainstem and spinal cord involvement caused by a novel mutation in the DARS2 gene. J Neurol. 2012;259:292-6.
- [Google Scholar]
- Spinal cord calcification in an early-onset progressive leukoencephalopathy. J Child Neurol. 2011;26:876-80.
- [Google Scholar]
- Leukoencephalopathy with brainstem and spinal cord involvement and lactate elevation (LBSL) with a novel DARS2 mutation and isolated progressive spastic paraparesis. J Neurol Sci. 2017;372:229-31.
- [Google Scholar]
- Extension of the phenotype of biallelic loss-of-function mutations in SLC25A46 to the severe form of pontocerebellar hypoplasia type I? Clin Genet. 2018;93:255-65. doi: 10.1111/cge.13084. Epub 2017 Nov 8.
- [Google Scholar]
- Loss of function of SLC25A46 causes lethal congenital pontocerebellar hypoplasia. Brain. 2016;139:2877-90.
- [Google Scholar]
- Infantile onset spinocerebellar ataxia is caused by recessive mutations in mitochondrial proteins twinkle and twinky. Hum Mol Genet. 2005;14:2981-90.
- [Google Scholar]
- Infantile onset spinocerebellar ataxia with sensory neuropathy (IOSCA): Neuropathological features. J Neurol Sci. 1998;161:57-65.
- [Google Scholar]
- Infantile-onset spinocerebellar ataxia: MR and CT findings. AJNR Am J Neuroradiol. 1995;16:1427-33.
- [Google Scholar]
- Mitochondrial recessive ataxia syndrome mimicking dominant spinocerebellar ataxia. J Neurol Sci. 2012;315:160-3.
- [Google Scholar]
- Leber's “plus”: Neurological abnormalities in patients with leber's hereditary optic neuropathy. J Neurol Neurosurg Psychiatry. 1995;59:160-4.
- [Google Scholar]
- Leber's hereditary optic neuropathy associated with a multiple-sclerosis-like picture in a man. Mult Scler. 2011;17:763-6.
- [Google Scholar]
- Acase of neuromyelitis optica harboring both anti-aquaporin-4 antibodies and a pathogenic mitochondrial DNA mutation for leber's hereditary optic neuropathy. Mult Scler. 2014;20:258-60.
- [Google Scholar]
- Severe inflammatory disease activity 14 months after cessation of natalizumab in a patient with leber's optic neuropathy and multiple sclerosis – A case report. BMC Neurol. 2016;16:197.
- [Google Scholar]
- C10ORF2 mutation associated with progressive external ophthalmoplegia and clinically isolated syndrome. Acta Neurol Belg. 2017;117:947-9.
- [Google Scholar]
- Magnetic resonance imaging correlates of genetically characterized patients with mitochondrial disorders: A study from South India. Mitochondrion. 2015;25:6-16.
- [Google Scholar]
- ISCA2 mutation causes infantile neurodegenerative mitochondrial disorder. J Med Genet. 2015;52:186-94.
- [Google Scholar]
- Early-onset cataracts, spastic paraparesis, and ataxia caused by a novel mitochondrial tRNAGlu (MT-TE) gene mutation causing severe complex I deficiency: A clinical, molecular, and neuropathologic study. J Neuropathol Exp Neurol. 2013;72:164-75.
- [Google Scholar]
- Acase of myelopathy, myopathy, peripheral neuropathy and subcortical grey matter degeneration associated with recessive compound heterozygous POLG1 mutations. Neuromuscul Disord. 2012;22:401-5.
- [Google Scholar]
- Alpha-synuclein pathology and parkinsonism associated with POLG1 mutations and multiple mitochondrial DNA deletions. Neuropathol Appl Neurobiol. 2009;35:120-4.
- [Google Scholar]
- Cerebro-spinal fluid findings in mitochondrial disorders. Mitochondrion. 2012;12:669-70.
- [Google Scholar]
- Abnormal evoked potentials of Kearns-Sayre syndrome. Electromyogr Clin Neurophysiol. 1995;35:365-70.
- [Google Scholar]
- Leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation 2010 May 25. 1993-2017. GeneReviews®. Seattle (WA): University of Washington, Seattle; Available from: http://www.ncbi.nlm.nih.gov/books/NBK43417
- [Google Scholar]
- Anesthesia for corrective spinal surgery in a patient with Leigh's disease. Anesth Analg. 2003;97:1539-41.
- [Google Scholar]
- Therapeutic strategies for mitochondrial disorders. Pediatr Neurol. 2015;52:302-13.
- [Google Scholar]




