
Translate this page into:
Homozygous germline c.3380C>G missense mutation in PNPLA6 gene in a case of Gordon Holmes syndrome associated with hypogonadotropic hypogonadism, cerebellar ataxia, and juvenile type tremor

*Corresponding author: Sezin Canbek, Department of Medical Genetics, Umraniye Research and Training Hospital, Umraniye, Istanbul, Turkey. sezin.canbek@ogr.iu.edu.tr
-
Received: ,
Accepted: ,
How to cite this article: Canbek S, Şenol M. Homozygous germline c.3380C>G missense mutation in PNPLA6 gene in a case of Gordon Holmes syndrome associated with hypogonadotropic hypogonadism, cerebellar ataxia, and juvenile type tremor. J Neurosci Rural Pract. 2024;15:594-9. doi: 10.25259/JNRP_24_2024
Abstract
Gordon Holmes syndrome (GDHS) is a genetic disorder that is inherited in an autosomal recessive manner. It is characterized by the presence of cerebellar ataxia, which refers to a lack of coordination and control of voluntary movements, and hypogonadotropic hypogonadism, which is a condition where the production of hormones that regulate sexual development and function is reduced. In this report, we describe the case of a Turkish patient who has been diagnosed with GDHS. The cause of this syndrome in the patient is a homozygous new mutation in the PNPLA6 gene. The proband case was detected through a collaboration between neurology and medical genetics based on her clinical symptoms. The specific point mutation was identified using the next-generation sequencing (NGS) technology. The patient, who was 28 years old, presented with primary amenorrhea, tremors in her head and both hands, cognitive impairment, cerebellar ataxia, hypogonadotropic hypogonadism, and diabetes. A point mutation, specifically a germline missense mutation c.3380C>G, was identified in exon 31 of the PNPLA6 (NM006702.5) gene. This gene is responsible for encoding the neuropathy target esterase protein. The mutation was found by NGS screening. Her parents were consanguineous and both heterozygous for the same missense mutation. This instance highlights the significant impact of first-degree consanguineous marriage on our nation, particularly in relation to autosomal recessive hereditary illnesses. It underscores the crucial function of genetic counseling in averting such scenarios. Subsequent findings of PNPLA6 variants will provide more elucidation on the correlation between patient genotype and phenotype. The finding of novel variations in every gene has been made feasible by the recent progress of genomic technology.
Keywords
Hypogonadotropic hypogonadism
Ataxia
Tremor
Germline c.3380C>G mutation
PNPLA6 gene
Chromosome 19p13.2
Gordon Holmes syndrome

INTRODUCTION
To learn more about the genetic causes of complex brain and metabolic diseases, our work is centered on an interesting case of a young woman who had signs that pointed to these types of syndromes.
Due to the patient’s uncertain initial diagnosis, we conducted an extensive clinical exome study using the next-generation sequencing (NGS) method for screening. A unique homozygous missense point mutation, namely, c.3380C>G (p.Ser1127Cys), was identified in the 31st exon of the PNPLA6 gene. This gene is recognized for its significant roles in neurological and metabolic processes. Further, clarification of this genetic variant provides insight into its potential ramifications in relation to documented clinical complaints. We provide a comprehensive assessment of the correlation between the identified genetic mutation and the intricate interplay between neurological and endocrinological observations in our patient.
Familial cerebellar ataxia and hypogonadism are uncommon conditions characterized by the presence of spinocerebellar atrophy together with hypogonadotropic hypogonadism.[1] The association between cerebellar ataxia and hypogonadism was first established in 1907, with evidence from post-mortem investigations.[2] In 1990, the first instance of cerebellar ataxia and non-neurological syndromes occurring together was reported. This case was likely caused by an autosomal recessive inheritance pattern and was named “congenital cerebellar hypoplasia associated with hypogonadotropic hypogonadism.” The consanguinity between the patient’s parents played a role in this condition. Due to the absence of a genetic test at that particular time, the patient’s condition was characterized only based on his clinical observations.[3]
There are two clinically recognized syndromes: Early-onset autosomal recessive cerebellar ataxias and hypogonadotropic hypogonadism. The first syndrome, known as I Boucher-Neuhäuser syndrome (MIM 215470), is also associated with chorioretinal dystrophy. This syndrome was first described by Boucher and Gibberd in 1969, and later by Neuhauser and Opitz in 1975 and Limber et al. in 1989.[4-7] The second syndrome, known as Gordon Holmes syndrome (GDHS) (MIM 212840), is also associated with brisk reflexes. This syndrome was first described by Holmes in 1907. The genetic etiology of many illnesses remains elusive despite multiple reports of familial inheritance. Moreover, it remains uncertain if these syndromes are distinct in terms of clinical and genetic characteristics, or whether they are instead groups of similar phenotypes along a continuum of neurodegenerative disorders caused by mutations in the same genes.[5,8] In this case, we present a young person who has a homozygous mutation and has tremor-predominant Boucher-Neuhäuser and GDHS. When evaluating children from families with consanguineous marriages, it is important to constantly consider the possibility of Boucher-Neuhäuser and GDHS. These syndromes are characterized by both growth retardation and delayed puberty, as well as the presence of tremors and neurodegenerative processes in later stages of life. The prevalence of tremor in previously documented cases was not highlighted. Due to the rarity of the condition and the possibility of phenotypic heterogeneity, the patient’s tremor and neurological diagnosis could not be established clinically and had to be determined by genetic testing.
For communities such as Turkey’s, where the proportion of consanguineous marriage is rather high, the importance of advanced molecular detection and genetic counseling in preventing autosomal recessive hereditary illnesses such as GDHS is highlighted in the current proband case.
CASE REPORT
This paper details the case of a 28-year-old female patient diagnosed with GDHS, which is characterized by a range of intricate clinical manifestations.
The patient is a graduate of a university who teaches in a rehabilitation facility. At the age of 9, she received a diagnosis of hypogonadotropic hypogonadism from the pediatric endocrinology department. This was due to her delayed growth, inability to acquire weight, and absence of menstruation at the age of 14. The measured levels of follicle-stimulating hormone, luteinizing hormone, and estradiol were also consistent with the diagnosis. The pelvic ultrasound revealed the uterus and ovaries in line with the patient’s age. The pituitary magnetic resonance imaging (MRI) conducted at that time did not reveal any abnormalities. The patient had hormone replacement treatment for low hormone levels, and menstruation was eventually established. The patient was continuing to use hormone replacement therapy. As a result, secondary sexual features also emerged.
At the age of 24, a significant imbalance in tremors became the main clinical symptom, resulting in the diagnosis of cerebellar ataxia. The neurological examination showed pronounced dysmetria on the right side, dysdiadochokinesia, poor heel-toe test, and a notable bilateral postural tremor on the right side. Brain MRI imaging could not rule out cerebellar atrophy, progressive degenerative processes, progressive supranuclear palsy, and cerebral abnormalities caused by hypoglycemia.
The patient’s weight was recorded as 49 kg and their height was assessed as 170 cm. It was established that her parents, who are first-degree relatives, were also carriers of the same mutation. The GDHS syndrome is characterized by many typical clinical manifestations, including cognitive impairment, primary amenorrhea, hypogonadotropic hypogonadism, cerebellar ataxia, and juvenile tremor. The investigation of the family’s genealogy showed that a certain kind of genetic mutation, known as a heterozygous germline missense point mutation, was inherited by family members. This mutation is found on a specific region of chromosome 19, known as 19p13.2. A crossover point mutation at codon 376 of exon 31 was found in both parents, who were totally healthy. The proband, on the other hand, had a homozygous missense point mutation [Figures 1-3].
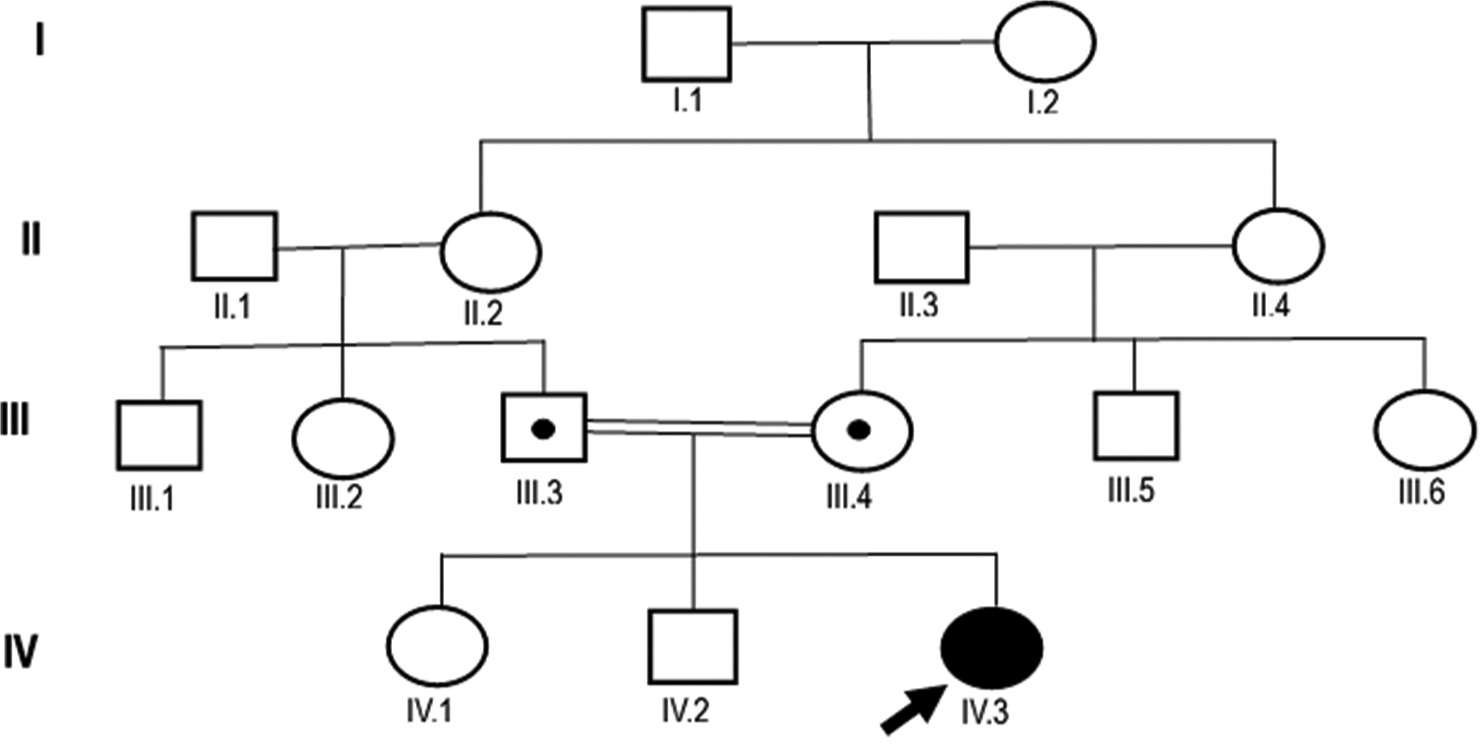
- Pedigree and clinical diagram of the presented proband case. The current female gordon holmes syndrome (GDHS) proband and her first-degree relative parents were evaluated clinically and for the target PNPLA6 gene. Family members are recognized by generation and number. The square indicates male family members; circle, female members; dotted circle and square indicate carrier individuals (parents) (III.3 and III.4). The arrow shows the proband case of GDHS (IV.3).
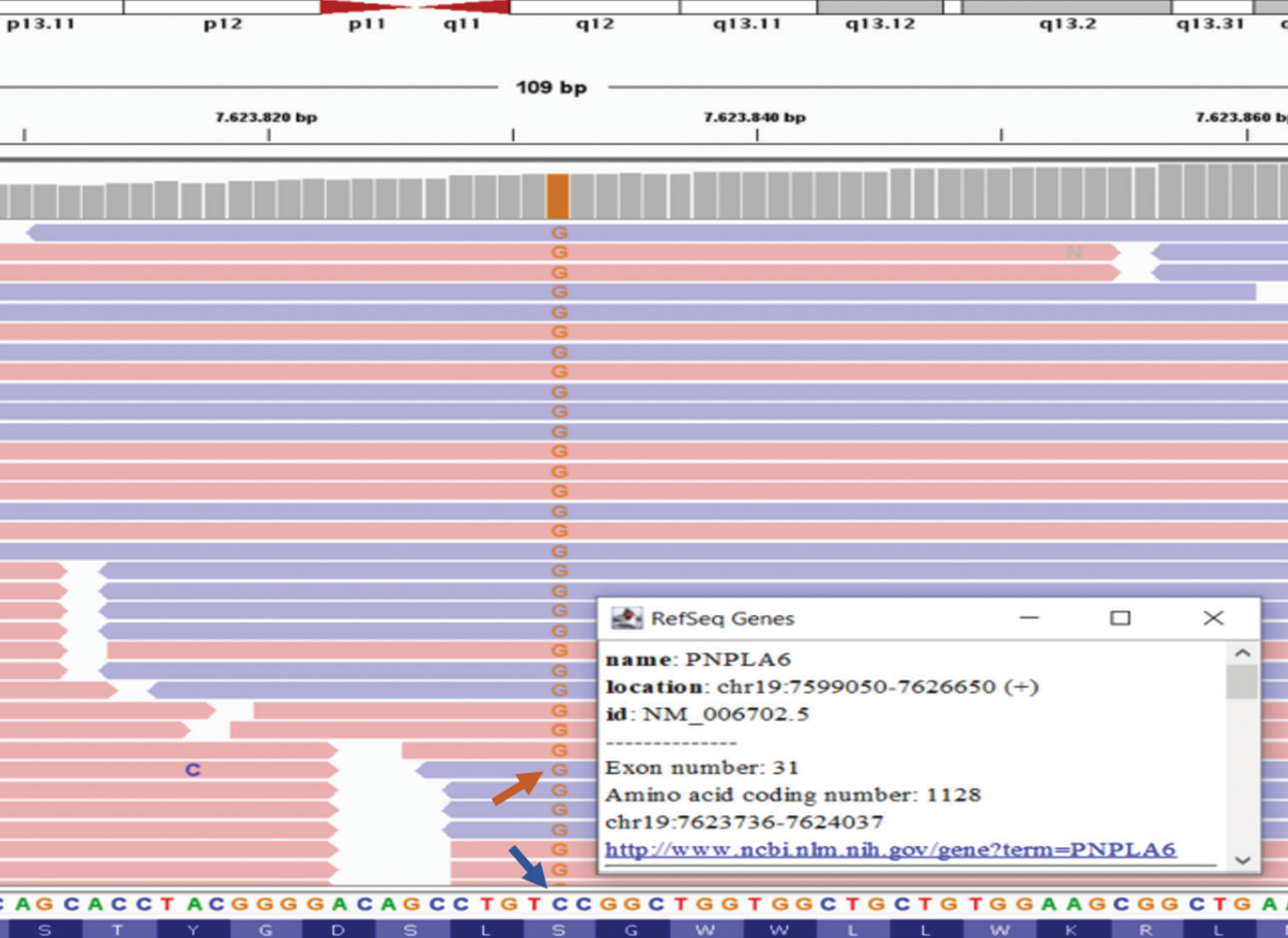
- The next-generation sequencing profile of the PNPLA6 (NM006702.5) gene for the proband case is depicted. In this particular case, we identified a homozygous missense point mutation, c.3380C>G (p.Ser1127Cys), in the 31st exon of PNPLA6. The blue line corresponds to the wild-type C base (normal), whereas the red arrow indicates the G base (homozygously mutated) at the transversion point, occurring at codon 376 of exon 31 for the PNPLA6 gene situated at the chromosome 19p13.2 locus.
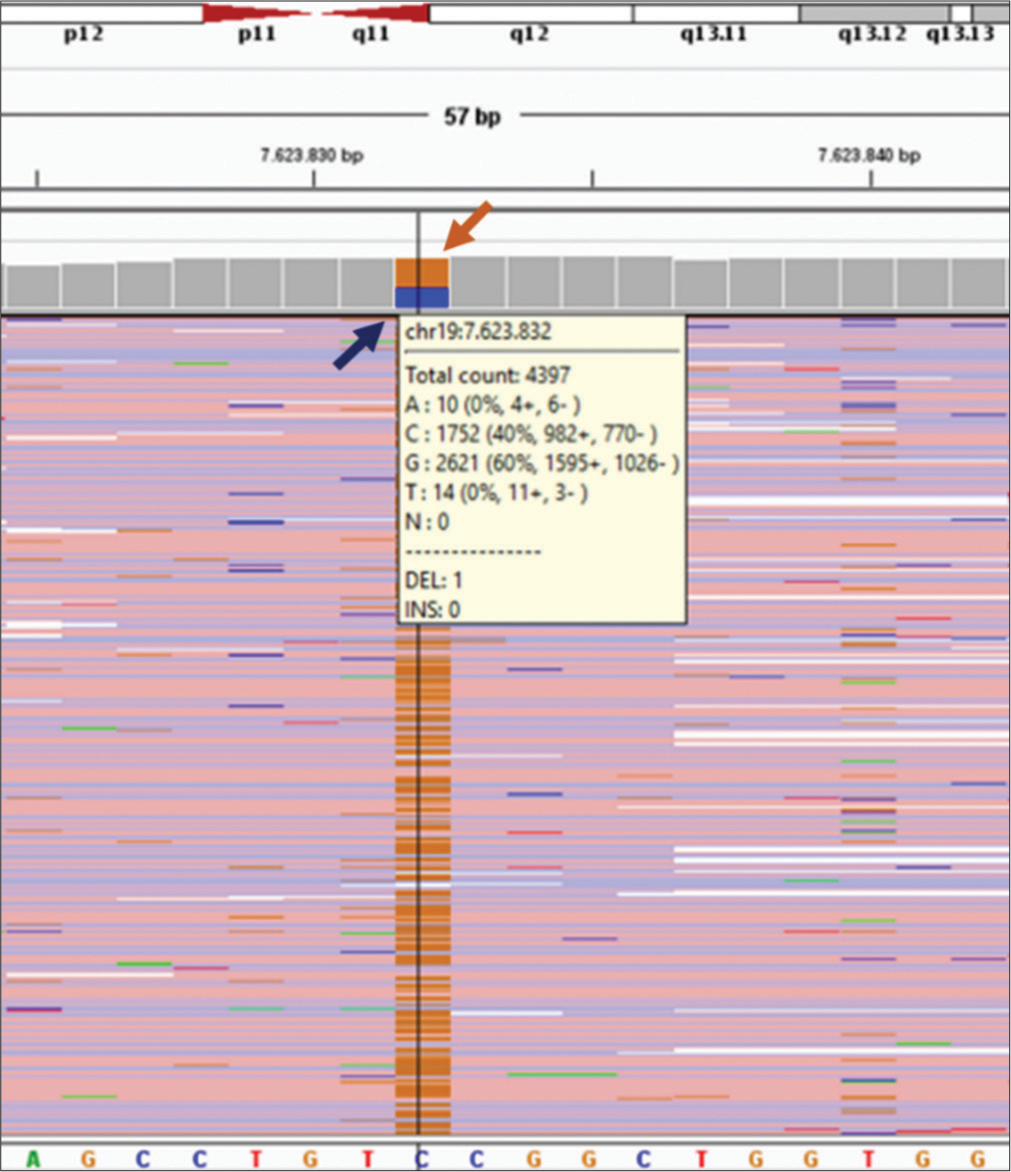
- The next-generation sequencing profile of the PNPLA6 (NM006702.5) gene for one of the parents (Father). Heterozygous missense point mutation of c.3380C>G (p.Ser1127Cys) in exon 31 of PNPLA6 was detected for his father, blue arrow indicates wild C base (normal) in 40% and the brown arrow indicates G base (heterozygous mutated). It shows 60%. Both parents were carriers for the transversional point mutation at codon 376 of exon 31 for the PNPLA6 gene located on chromosome 19p13.2 for the current proband case.
Diagnostic approaches and methodology
A collaborative effort between neurology and medical genetics was required for the multidisciplinary diagnosis, with a particular emphasis placed on clinical findings.
The identification of a germline missense point mutation (c.3380C>G) in exon 31 of the PNPLA6 (NM006702.5) gene, which is responsible for generating the neuropathy target esterase (NTE) protein, was made possible by the use of NGS methods.
In summary, the laboratory procedure consists of amplifying the specific gene area(s) linked to the illness by polymerase chain reaction and then sequencing this region using NGS technology. The Clinical Exome Solution by Sophia Genetics kit is used for this specific objective. The sequencing reaction is performed with the Illumina NextSeq® equipment and reagent kits that are compatible with it. Raw data was analyzed using the Sophia DDM® data analysis platform as part of the data analysis process. Pepper®, a proprietary baseline method from Sophia Genetics, was used to conduct alignment and variant identification against the hg19 human genome reference.
The process of variant annotation was carried out using Sophia Genetics’ MOKA® software. For each variant, the impact on the protein sequence (such as missense or stop gain), the frequency of occurrence in different populations (1000G, ESP, ExAC, gnomAD), and the predictions made by algorithms such as sorting intolerant from tolerant (SIFT) and PolyPhen were determined. Details on the detrimental impact of the variant have been included. The identification of copy number variations was carried out using the MUSKAT® program developed by Sophia Genetics.
The TruSight One Expanded Illumina panel enabled gene profiling, while the NGS technology enabled precise diagnosis and correlation between particular genotypes and phenotypes. A thorough assessment was carried out using the Sophia DDM platform, which integrated data from gnomAD, ClinVar, dbSNP, and the 1000 Genome datasets.
The use of this comprehensive method facilitated the clinical examination of the present individual’s exome, resulting in a thorough comprehension of the genetic makeup. The NGS findings were validated by Sanger analysis for both the patient and her relatives. It was ascertained that both parents were carriers.
Overall, the genetic test conducted on the patient revealed that the identified missense mutation was homozygous. The variation is considered to be new (PM2) as it has not been seen in the 1000 Genomes Project, ESP5400, and GnomAD databases. In silico analyses suggest that the variation is likely to have a deleterious impact on the protein (PP3). When a variation is assessed using the American college of medical genetics (ACMG) evidence, it is categorized as a variant of uncertain (or unknown) significance (VOUS).
DISCUSSION
Clinicians have a twofold difficulty when dealing with young-onset cerebellar diseases since they are both intriguing and demanding. Clinicians who treat patients with cerebellar ataxia need to have a deep grasp of both common and unusual causes of the condition to effectively diagnose and treat it. Cerebellar ataxia is often seen in neurology wards among younger individuals. This disorder may be caused by several circumstances, including infections, drug toxicity, metabolic problems, space-occupying lesions, genetic illnesses, and others.[9,10] Boucher-Neuhauser disease (BNHS) is a rare clinical condition associated with various syndromes. It is characterized by the early development of ataxia, hypogonadism, and chorioretinal degeneration. The disease is inherited through autosomal recessive genes.[5]
This paper investigates a fascinating instance of ataxia with tremor, revealing a homozygous variant in the PNPLA6 gene. The PNPLA6 gene plays a crucial role in this story by encoding NTE, a membrane enzyme that is essential for cellular signaling and neural development.[8,11] Originally connected with SPG39 and defects in motor neurons,[12] further studies broadened its range of consequences, establishing a connection between PNPLA6 mutations and different diseases.[13] The mutations exhibit a range of clinical characteristics, such as cerebellar ataxia, involvement of the higher motor neurons, hypogonadotropic hypogonadism, chorioretinal degeneration, peripheral neuropathy, hair abnormalities, and cognitive impairment. The clinical disorders that arise as a consequence are known as BNHS and GDHS.[8]
The patient being observed had a notable combination of symptoms related to the cerebellum, including unsteady trunk movements, involuntary eye movements, decreased muscle tone, exaggerated reflexes, trouble with rapid alternating movements, and speech impairment. These symptoms together suggest the presence of cerebellar ataxia. Electromyography provided evidence to support this diagnosis by detecting neuropathy. Moreover, the patient’s delayed sexual development indicated the presence of hypogonadotropic hypogonadism. An extensive multidisciplinary assessment resulted in the diagnosis of GDHS. The NGS has revealed a germline missense mutation in exon 31 of the PNPLA6 (NM006702.5) gene (c.3380C>G), making genetic testing a useful tool. Interestingly, the patient’s parents, as first-degree relatives, both carried the same genetic mutation.
Aside from the complex medical details, this case reveals a wider story concerning the misleading consequences of marriages between close relatives, especially in relation to genetic illnesses inherited in an autosomal recessive manner, such as GDHS. Genetic counseling has a key role in reducing such situations, providing an essential preventative strategy. The discovery of genetic alterations related to consanguinity highlights the need to raise awareness about possible hazards and provide support for informed family planning choices.
CONCLUSION
The very complex interaction between genetics and clinical symptoms that was shown in this GDHS case highlights the need to take a holistic and multidisciplinary approach to the diagnosis and treatment of uncommon cerebellar illnesses. Not only does having a comprehensive knowledge of the genetic basis make it easier to make accurate diagnoses but it also highlights how important it is to intervene at an early stage. The need for genetic counseling becomes even more apparent when it comes to preventing situations like these and encouraging individuals to make well-informed choices on family planning.
This instance has implications that extend beyond the specific patient, shedding light on the broader social consequences of genetic abnormalities caused by consanguinity. The crucial role of education and counseling in reducing the incidence of such diseases is evident. This case highlights the importance of using a thorough approach that combines genetic knowledge to understand the intricate nature of rare cerebellar disorders in young people. This approach is crucial not only for the well-being of patients but also for gaining a broader understanding of and preventing genetic conditions associated with consanguinity.
Acknowledgements
The authors thank the family members who agreed to participate in the presented results.
Data availability statement
All data generated or analyzed during this study are included in this published article.
Authors’ contributions
SC and MGŞ: Acquisition of data, analyzed the clinical data, and designed the clinical experiments, SC: Interpretation of data, analyzed the NGS screening, wrote the manuscript, supervised the study, and reviewed the manuscript. Both the authors read and approved the final manuscript.
Ethical approval
The Institutional Review Board approval is not required.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship
Nil.
References
- Familial cerebellar ataxia and hypogonadotropic hypogonadism: Evidence for hypothalamic LHRH deficiency. J Neurol Neurosurg Psychiatry. 1982;45:747-51.
- [CrossRef] [PubMed] [Google Scholar]
- A form of familial degeneration of the cerebellum. Brain. 1908;30:466-89.
- [CrossRef] [Google Scholar]
- Congenital cerebellar hypoplasia and hypogonadotropic hypogonadism. J Neurol Sci. 1990;98:259-65.
- [CrossRef] [PubMed] [Google Scholar]
- Familial ataxia, hypogonadism and retinal degeneration. Acta Neurol Scand. 1969;45:507-10.
- [CrossRef] [Google Scholar]
- Boucher neuhauser syndrome-a rare cause of inherited hypogonadotropic hypogonadism. A case of two adult siblings with two novel mutations in PNPLA6. Eur J Med Genet. 2017;60:105-9.
- [CrossRef] [PubMed] [Google Scholar]
- Autosomal recessive syndrome of cerebellar ataxia and hypogonadotropic hypogonadism. Clin Genet. 1975;7:426-34.
- [CrossRef] [PubMed] [Google Scholar]
- Spinocerebellar ataxia, hypogonadotropic hypogonadism, and choroidal dystrophy (Boucher-Neuhauser syndrome) Am J Med Genet. 1989;33:409-14.
- [CrossRef] [PubMed] [Google Scholar]
- PNPLA6 mutations cause Boucher-Neuhauser and Gordon Holmes syndromes as part of a broad neurodegenerative spectrum. Brain. 2014;137(Pt 1):69-77.
- [CrossRef] [PubMed] [Google Scholar]
- Rare case of Gordon Holmes syndrome. BMJ Case Rep. 2018;2018:bcr2018225638.
- [CrossRef] [PubMed] [Google Scholar]
- Hypogonadotropic hypogonadism and cerebellar ataxia: Detailed phenotypic characterization of a large, extended kindred. J Clin Endocrinol Metab. 2002;87:1607-12.
- [CrossRef] [PubMed] [Google Scholar]
- Roles of NTE protein and encoding gene in development and neurodevelopmental toxicity. Chem Biol Interact. 2016;259(Pt B):352-7.
- [CrossRef] [PubMed] [Google Scholar]
- Neuropathy target esterase gene mutations cause motor neuron disease. Am J Hum Genet. 2008;82:780-5.
- [CrossRef] [PubMed] [Google Scholar]
- Oliver McFarlane syndrome: Two new cases and a review of the literature. Ophthalmic Genet. 2021;42:464-73.
- [CrossRef] [PubMed] [Google Scholar]




