
Translate this page into:
Gliomatosis cerebri: Case series of six cases with review of literature
Address for correspondence: Dr. Vamsi Krishna Yerramneni, Department of Neurosurgery, Nizam's Institute of Medical Sciences, Punjagutta, Hyderabad - 500 082, Telangana, India. E-mail: vamsiky.ns@gmail.com
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Background:
Gliomatosis cerebri is characterized by diffuse infiltration of glial cells with preservation of neuronal architecture. It is an uncommon glial neoplasm of astrocytic origin that occurs in adults and is exceedingly rare in children.
Materials and Methods:
The authors retrospectively analyzed the data of 6 patients of gliomatosis cerebri operated between 2007 and 2012.
Result:
All patients underwent surgical decompression, followed by chemoradiotherapy. The survival ranged between 3 and 45 months. The mean survival was 18.5 years.
Conclusion:
Performance scores at presentation and the nonglioblastomatous histology seems to favorably affect the prognosis. Larger studies are required to comment on the role of combination of surgery, chemoradiotherapy as a treatment modality.
Keywords
Chemoradiotherapy
gliomatosis cerebri
prognosis

Introduction
Gliomatosis cerebri is characterized by diffuse infiltration of glial cells with preservation of neuronal architecture. The term gliomatosis cerebri was first introduced by Nevin in 1938.[1] It is an uncommon glial neoplasm of astrocytic origin that occurs in adults and is exceedingly rare in children.[1] It is classified as Grade III astrocytoma according to 2007 World Health Organization classification of central nervous system (CNS) tumors. Different chemotherapeutic agents and radiotherapy regimens have been used, but the prognosis still remains poor. The available literature suggests age, male sex, performance scores at the presentation, and site of involvement in magnetic resonance imaging (MRI) as the prognostic factors.[2] The authors studied different prognostic factors and the outcomes with temozolomide and radiotherapy in six cases of gliomatosis cerebri treated at their institution from 2007 to 2012.
Materials and Methods
Six patients treated at Department of Neurosurgery, Nizam's Institute of Medical Science from July 2007 to February 2012 with a diagnosis of gliomatosis cerebri were included in the study. Data were obtained by reviewing the admission case files, radiation oncology, radiology, and pathology records. The radiological criteria employed were widespread involvement of white matter with preservation of normal anatomical architecture in two or more lobes of the brain. All patients underwent clinical examination followed by MRI brain plain and contrast imaging studies on admission. Surgery was done in all patients. Surgical plan varied based on the location of the tumor. Since the tumor is of diffusely infiltrative nature, the aim of surgery in all patients was subtotal excision. Postoperatively, 2 patients received concurrent chemoradiotherapy followed by adjuvant temozolomide, 2 patients received radiotherapy alone. Follow-up data were obtained from follow-up visits and telephonic interviews. A retrospective analysis was performed using the clinical charts, radiological investigations, and pathological examinations.
Results
Age of the patients at diagnosis ranged from 16 to 60 years, with mean age was 41.3 years, median age was 42 years. The male:female ratio was 2:1. The duration of symptom before diagnosis ranged from 2 days to 10 years. The range of karnofsky score at the time of presentation was 30–90. Presenting symptoms included headache, seizures, and altered sensorium. Two patients presented with hemiparesis. Examination findings included papilledema in two cases, cranioneuropathy in 4 patients, upper motor neuron type of facial palsy, impaired gait disturbances and impaired extraocular movements in 1 patient each. The clinical features of the 6 patients are summarized in [Table 1].
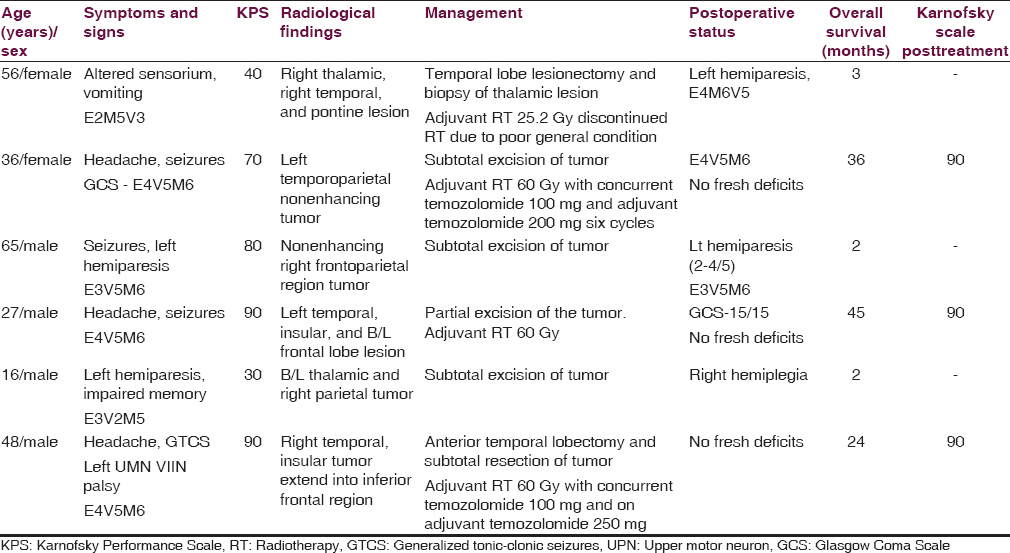
Radiological findings
All 6 patients underwent computed tomography (CT) and MRI of the brain. CT showed poorly defined low-density lesions in four cases. In two cases, CT revealed obliteration of lateral ventricle and basal cisterns with midline shift suggestive of increased intracranial pressure. Patchy light contrast enhancement was seen only in one case. MRI T1- and T2-sequences showed Temporo-parietal lobes involvement predominantly with temporal lobe involvement seen in 4 patients, parietal lobe involvement in three, and frontal lobe involvement in 2 patients [Figure 1]. In addition to the lobes involvement, insula involvement was seen in 2 patients. Two patients had bilateral thalamic and frontal lobe involvement. Supra and infratentorial hyperintensities were seen in 2 patients. Contrast enhancement is hardly seen with only two cases showing mild patchy enhancement of areas corresponding to the hyperintense areas on T2-sequence.
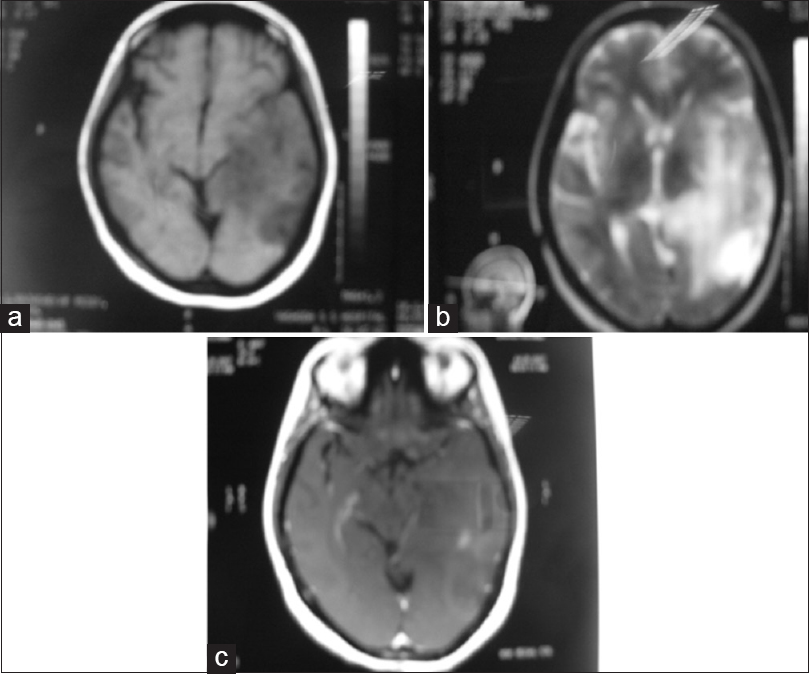
- (a) T1-weighed magnetic resonance imaging brain showing ill-defined hypointense lesion. (b) T2-weighted magnetic resonance imaging showed an area of diffuse poorly defined high signal intensity with a variable degree of obliteration of sulci and gyri. (c) Contrast magnetic resonance imaging shows mild diffuse patchy enhancement
Histological features
The histology from resected specimens was essentially similar. The cells were small with elongated nuclei; the cytoplasm was scant to moderate with few gemistocytes. There was nuclear pleomorphism [Figure 2a and b]. The mitosis was few; the Ki67 ranged from 3% to 10% and GFAP was positive in all cases [Figure 3]. There was no microvascular proliferation orarea of necrosis.
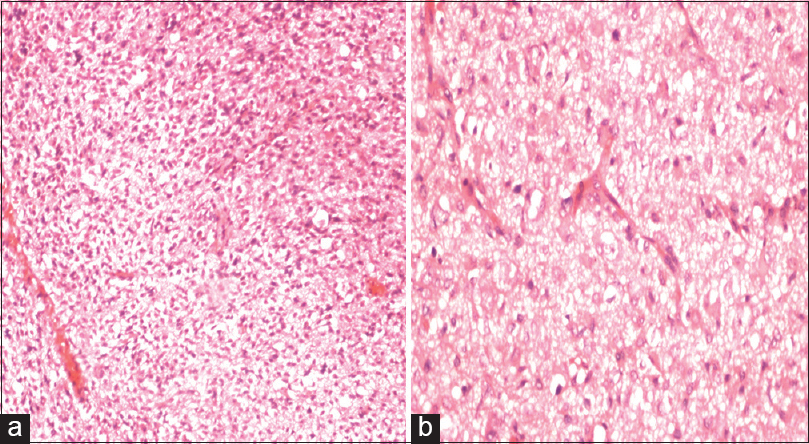
- (a) H and E ×10 diffusely infiltrating small round to oval cells with pleomorphic nuclei. (b) H and E ×40 gemistocytic astrocytes and scattered mitotic figures
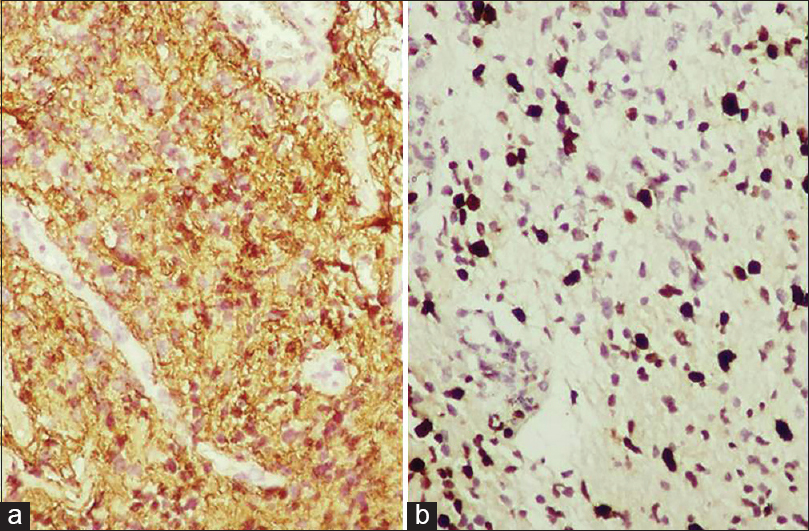
- Immunohistochemistry of tumour cells showing GFAP positivity, Ki 67 labelling index- 40%
Treatment and outcome
All 6 patients underwent surgery as the primary modality of treatment. Five patients underwent craniotomy and subtotal excision of the tumor. Only biopsy was performed in one case as the lesion was too diffuse. Emergency decompression was done in 1 patient due to sudden deterioration of Glasgow Coma Scale. Postoperative recovery was uneventful in all cases. There was no operative mortality. Adjuvant radiotherapy protocol consisted of radiotherapy to preoperative T2 fluid-attenuated inversion recovery (FLAIR) edema with margin to a total dose of 60 Gy, 2 Gy per fraction, and 30 fractions. Concurrent chemotherapy was given with temozolomide 75 mg/m2 followed by adjuvant temozolomide 150 mg/m2 every 28 days for 6 cycles. Postoperative concurrent chemoradiotherapy, followed by adjuvant chemotherapy was given in two cases; radiotherapy only was given in two cases. One patient developed subacute intestinal obstruction, renal failure, discontinued radiotherapy and expired after 2 months (case 1), another patient survived 3 years, and 2 patients improved symptomatically and are on follow-up. Two patients with poor Karnofsky Performance Scale score did not receive adjuvant treatment and survived 2 months (case 3, 5). Two patients did not show progression at follow up of 2 years, overall survival ranged from 2 to 36 months, median overall survival was 4.5 months, mean overall survival was 18.5 months.
Discussion
Gliomatosis cerebri is a rare brain tumor and it is often difficult to establish a definitive diagnosis. Before the era of CT, the diagnosis of most reported cases of gliomatosis cerebri was based on autopsy findings. The classical diagnostic criteria proposed by Scheinker and Evans in 1943 were based upon autopsy findings of widespread infiltration of neoplastic glial cells. A diagnostic criterion proposed by Kleihues et al. was the involvement of at least two lobes of the brain by small elongated cells without a cellular, centrally necrotic center.[3] Burger and Schithaur defined gliomatosis cerebri as a diffuse infiltrating glioma, which may involve the supratentorial compartment, posterior fossa, or even intraspinal parenchyma, sometimes in continuity.[4]
Male sex appears to be affected more in most of the series as is the case in our series also.[235] The disease occurred in all age groups but was most common in the fifth decade with a second peak in the second decade similar to the trend seen in our limited series.[6] Clinical presentation is subtle and nonspecific, reflects the diffuse neuronal disruption caused by the disease manifesting as seizures, focal neurological deficits, memory loss, and other higher mental functions impairment.[5] Radiological diagnosis is difficult in view of the nonspecific features. CT may either appear normal or shows subtle squashing of the ventricles or hypodensity or diffuse cerebral edema resulting in obliteration of the basal cisterns and midline shift. MRI brain T2-weighted images and FLAIR images show heterogenous hyperintensities appearing similar to CNS infiammatory diseases, vasculitis, leukoencephalopathies or venous infarcts. Nevertheless, a diagnosis of GC must be entertained in cases showing MRI T2 and FLAIR heterogenous hyperintensities with mass effect, loss of gray-white differentiation, and no uptake of contrast. The relative preservation of the blood-brain barrier is thought to be the reason for nonenhancement of contrast. In many instances, the diagnosis of GC has been delayed, because of the rarity and difficulty of diagnosis.[7]
Histologically, tumor cells diffusely infiltrate white and gray matter and sometimes exhibited the perineuronal, perivascular or subpial tumor cell collections. The infiltrating cells showed variation from small cells with little cytoplasm to cells with moderate cytoplasm and the nuclei from elongated, oval to round. There was a wide variety of cellularity and cytological composition showing their malignant nature.[3] Angiogenesis was absent in the gliomatosis cerebri, suggesting that tumor growth in these neoplasms is supported by coaptation of the existing vasculature and not by the formation of new vessels.[8] The real pathological lesion demonstrated at autopsy is always wider than the lesion shown in MRI or positron emission tomography.[3] Morphologically, tumors were identified as astrocytic, oligodendroglial, and mixed with astrocytic variety being common.[910] Oligodendroglial variant is more common in males and has a better prognosis while the astrocytic variant carries worst prognosis. Median survival is least in astrocytic tumors around 11 months, and the oligodendroglial variant has survival up to 36 months.[2]
The study done by authors being retrospective with small sample size has inherent limitations in predicting and assessing the influence of various factors on survival. In our series, the median survival is 18.5 months, near to in the ANOCEF database of 14.5 months.[2] In our series, 3 patients with good performance score has survival ranging from 6 months to 36 months. Poor performance scores at presentation have uniformly predicted bad prognosis even in the presence of favorable histology.
The molecular basis is unknown, a number of reports demonstrated molecular alterations similar to those seen in common gliomas.[9] These studies also suggested that gliomatosis cerebri seems to represent a monoclonal rather than a polyclonal process.
The treatment of GC is controversial. Most of the literature available so far is based on the retrospective studies. All the reports summarize various combinations of radio and chemotherapies.[231011] Elshaikh et al. presented 8 of 12 patients who received only radiation treatment (median dose of 55.4 Gy).[11] The clinical and radiological findings improved in 3 patients, stabilized in 3 patients, and deteriorated in 2 patients. Median survival was 11.4 months, and the authors concluded that radiation alone was not sufficient, and, therefore, more aggressive therapy may be needed. Perkins et al. in their retrospective study of 30 patients concluded that RT is effective against gliomatosis cerebri. Patients who are young and have a nonglioblastoma tumor of histologic subtype perform more favorably. Median survival was 18 months.[12] Though in the aforementioned studies, only radiotherapy alone was used, the authors expressed contradicting views regarding the radiotherapy as the only treatment modality.
There are studies using chemotherapy as the only modalities showing comparable outcome with the series using radiotherapy only.[51314] Levin et al. in their study of 11 patients on initial chemotherapy having disease progression-free survival of 12 months concluded that temozolomide chemotherapy can be used initially, and radiotherapy can be deferred until the disease progression is documented[13] Taillibert et al. in their analysis of 296 individual cases including 90 patients followed through the ANOCEF network, and 206 cases from the literature concluded that despite a high rate of stabilization, the survival impact of WBRT, which carries the risk of severe toxicity, remains unclear.[2]
From the current study, it can be observed that the good performance score at presentation definitely has a survival more than 6 months. Surgical decompression with chemoradiotherapy only seem to have some effect in controlling the disease progression in patients having a good performance score at presentation does not seem to have affected the prognosis. Surgery does have a definite role in patients presenting with raised intracranial pressure. Apart from this, the nonglioblastoma type histology coupled with good performance scores at presentation seems to have favorable outcomes.
Conclusion
The glomatosis cerebri, an aggressive tumor with a resemblance to many neurological diseases at presentation. The overall prognosis is poor. Adjuvant radiotherapy and chemotherapy and nonglioblastoma type histology seem to have favorable outcomes only in cases presenting with a good performance score at presentation.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Diffuse cerebrospinal gliomatosis: Clinical and radiological findings. Br J Neurosurg. 1988;2:415-8.
- [Google Scholar]
- Gliomatosis cerebri: A review of 296 cases from the ANOCEF database and the literature. J Neurooncol. 2006;76:201-5.
- [Google Scholar]
- Gliomatosis cerebri: Clinical features, treatment, and prognosis. Acta Neurochir (Wien). 1998;140:755-62.
- [Google Scholar]
- Gliomatosis cerebri – An appropriate diagnosis?. Case reports. Acta Radiol. 1997;38:381-90.
- [Google Scholar]
- Gliomatosis cerebri: A clinical, radiological and pathological report of four cases. Br J Neurosurg. 1991;5:187-93.
- [Google Scholar]
- Gliomatosis cerebri mimicking Rasmussen encephalitis. Case report. J Neurosurg. 2007;107:143-6.
- [Google Scholar]
- Gliomatosis cerebri: Quantitative proof of vessel recruitment by cooptation instead of angiogenesis. J Neurosurg. 2005;103:702-6.
- [Google Scholar]
- Gliomatosis cerebri: Molecular pathology and clinical course. Ann Neurol. 2002;52:390-9.
- [Google Scholar]
- Combined surgery, radiation, and chemotherapy for oligodendroglial gliomatosis cerebri. Br J Neurosurg. 2004;18:306-10.
- [Google Scholar]
- Gliomatosis cerebri: Treatment results with radiotherapy alone. Cancer. 2002;95:2027-31.
- [Google Scholar]
- Gliomatosis cerebri: Improved outcome with radiotherapy. Int J Radiat Oncol Biol Phys. 2003;56:1137-46.
- [Google Scholar]
- Chemotherapy as initial treatment in gliomatosis cerebri: Results with temozolomide. Neurology. 2004;63:354-6.
- [Google Scholar]




