
Translate this page into:
Extensive Cerebral Cortical Involvement in Aquaporin 4 Antibody Positive Neuromyelitis Optica Spectrum Disorder
Kiran Kumar Ramineni, MD, DM, Consultant Neurologist Department of Neurology Yashoda Superspeciality Hospital, Near Nalgonda Cross Roads, Malakpet, Hyderabad, 500036, Telangana India ram.kirankumar.dm@gmail.com
This article was originally published by Thieme Medical and Scientific Publishers Pvt. Ltd. and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Abstract
Neuromyelitis optica spectrum disorders (NMOSDs) usually manifest with features of long-segment myelitis and/or optic neuritis. Area postrema involvement presents with nausea, vomiting, and intractable hiccups. Cerebral cortical involvement is a relatively uncommon phenomenon. This report describes an interesting case of aquaporin 4 antibody positive NMOSD with extensive cerebral cortical involvement in addition to area postrema and cervicomedullary lesions observed on magnetic resonance imaging. Following immunosuppressive therapy, good clinical response and near-complete resolution of brain imaging abnormalities were observed.
Keywords
aquaporin 4
cerebral cortex
NMOSD
magnetic resonance imaging

Introduction
Neuromyelitis optica is an autoimmune neurological disorder with recurrent attacks of myelitis and optic neuritis. Development and availability of accurate assays for the detection of specific aquaporin 4 antibody (Nmo-IgG) helped in the conceptualization of much broader term, neuromyelitis optica spectrum disorder (NMOSD).1 Although various atypical clinical and imaging findings of NMOSD are reported, significant cerebral cortical involvement is extremely uncommon. We report an interesting case of aquaporin 4 antibody positive NMOSD with extensive cerebral cortical involvement as the likely underlying pathogenesis for vision impairment.
Case Report
A 21-year-old lady was brought to our hospital with history of blurring of vision for the last 5 days. She had mild holocranial headache but no history of diplopia or periorbital pain. She developed severe nausea with recurrent vomiting and hiccups for the last 4 days. One day before, she developed one episode of generalized tonic clonic seizure. She had no history of fever, rash, joint pains, or recent vaccination. She did not have any significant past neurological illness, had normal birth, and development and family history were noncontributory.
She was drowsy, confused, and irritable. Vitals were as follows: temperature, 99 F; pulse rate, 110/min regular; blood pressure, 100/70 mmHg; and respiratory rate, 28/min. Fundus examination was normal, relative afferent pupillary defect was absent, and pupils were 3 mm in both eyes equally reacting to light. She was able to perceive light and hand movements very close to the face. No paucity of limb movements while on bed, deep tendon reflexes 2+, plantar withdrawals, and no objective sensory loss or level were noted. She had no bowel or bladder disturbances but had terminal neck stiffness. In the emergency department 1,500 mg of intravenous levetiracetam was administered, and then she was shifted to neuro intensive care unit.
Contrast-enhanced magnetic resonance imaging (MRI) of brain revealed T2, FLAIR—fluid attenuated inversion recovery—hyperintensities in bilateral parieto-occipital regions with significant cortical involvement; area postrema and cervicomedullary regions with enhancement; and diffuse leptomeningeal enhancement with normal appearing optic nerves (Fig. 1). Possibilities considered were atypical presentation of NMOSD or acute disseminated encephalomyelitis (ADEM).
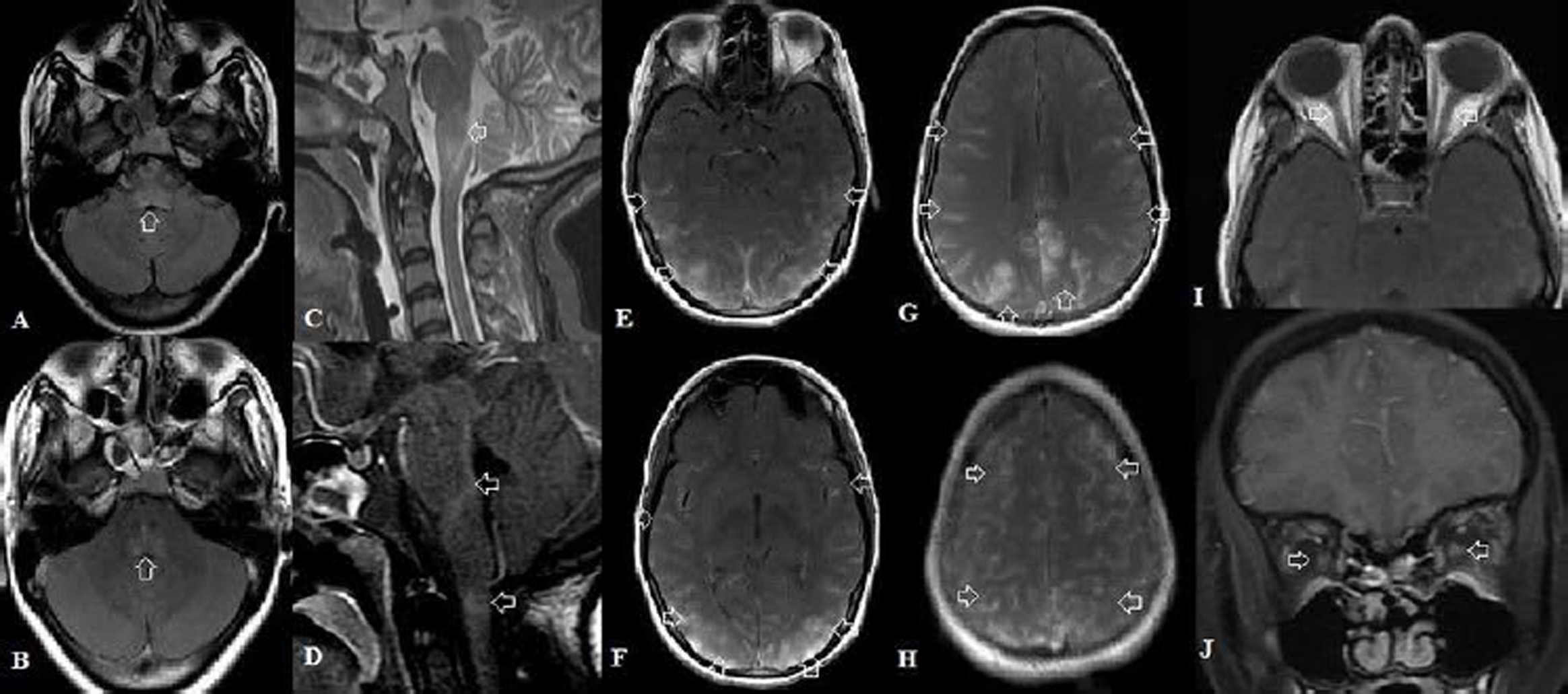
-
Fig. 1 (A) Axial FLAIR—fluid attenuated inversion recovery—and (C) sagittal STIR—short TI inversion recovery—and corresponding axial and sagittal postcontrast images. Images (B) and (D) show hyperintense signal in the area postrema, with postcontrast enhancement in area postrema and cervicomedullary junction. Axial postcontrast images (E–H) show diffuse cortical and leptomeningeal enhancement in bilateral cerebral hemispheres, more marked in parieto-occipital regions. Axial (I) and coronal (J) postcontrast images show normal optic nerves with no postcontrast enhancement.
Fig. 1 (A) Axial FLAIR—fluid attenuated inversion recovery—and (C) sagittal STIR—short TI inversion recovery—and corresponding axial and sagittal postcontrast images. Images (B) and (D) show hyperintense signal in the area postrema, with postcontrast enhancement in area postrema and cervicomedullary junction. Axial postcontrast images (E–H) show diffuse cortical and leptomeningeal enhancement in bilateral cerebral hemispheres, more marked in parieto-occipital regions. Axial (I) and coronal (J) postcontrast images show normal optic nerves with no postcontrast enhancement.
Cerebrospinal fluid analysis revealed 6 cells/mm3, and normal protein, sugar, and adenosine deaminase levels. Meningoencephalitis panel, including herpes simplex virus 1 and 2 polymerase chain reaction, was negative. Routine laboratory investigations, including complete blood picture, erythrocyte sedimentation rate, glycosylated hemoglobin, serum electrolytes, creatinine, serum calcium, and liver function tests, were within normal range. Chest X-ray, electrocardiogram, and ultrasound abdomen were unremarkable.
Serum aquaporin 4 antibody (Nmo-IgG) was positive and myelin oligodendrocyte glycoprotein (MOG) antibody was not detected. Rheumatoid factor, antinuclear antibody profile, and serum angiotensin converting enzyme levels were within normal limits. Antineutrophil cytoplasmic antibody and human immunodeficiency virus 1 and 2 were negative. Final diagnosis of aquaporin 4 antibody positive NMOSD with extensive cerebral cortical involvement along with area postrema syndrome was made.
Intravenous levetiracetam 500 mg twice a day was continued and methyl prednisolone 1 g daily was started for 5 days along with symptomatic medication and supportive care. She had significant clinical improvement over next 10 days. Oral prednisolone was tapered slowly over next few months and azathioprine dose titrated gradually.
MRI brain repeated after 4 months showed significant resolution of initial abnormalities (Fig. 2). She maintained good clinical improvement and did not have any relapse over next 18 months.
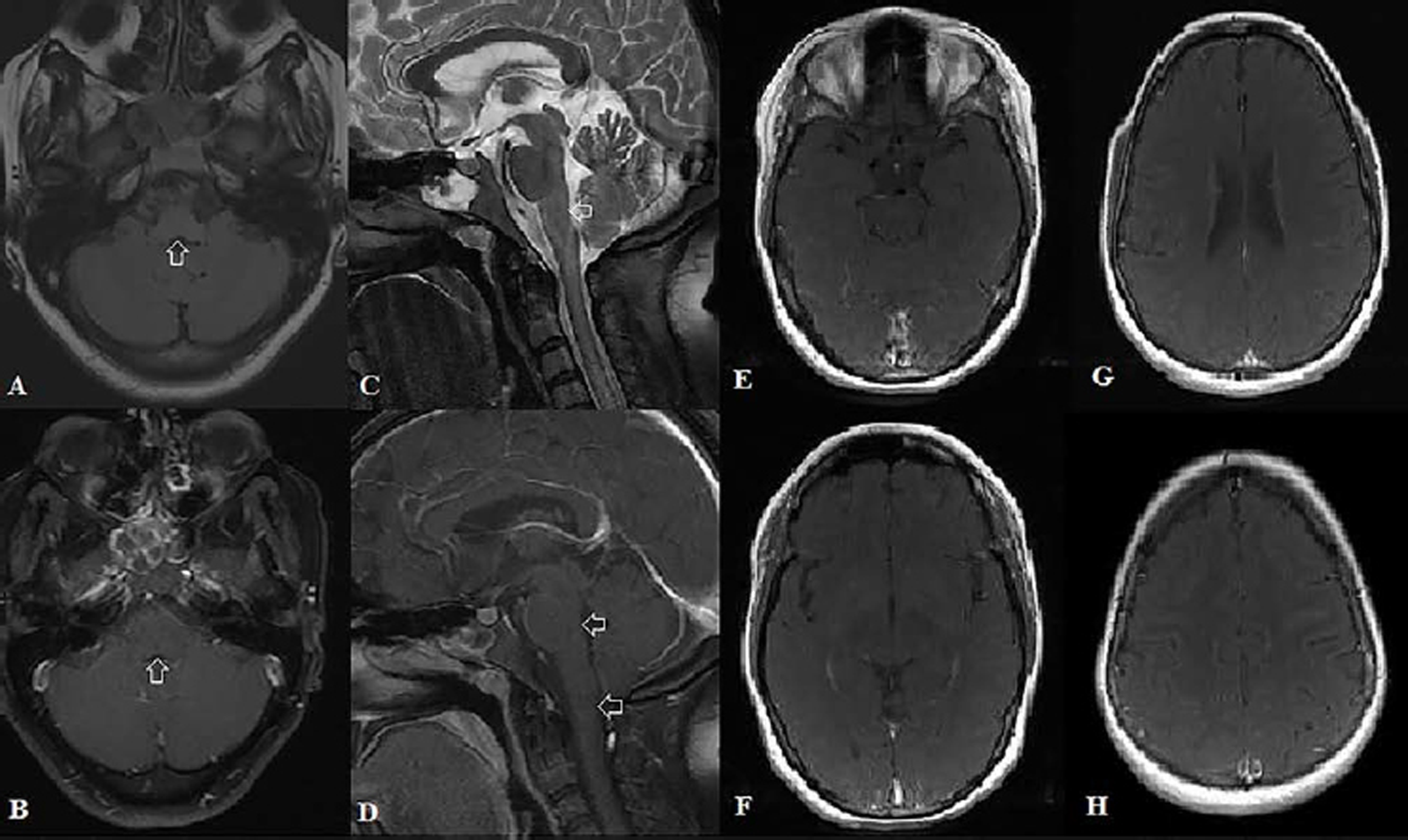
-
Fig. 2 Follow-up magnetic resonance imaging. (A) Axial FLAIR—fluid attenuated inversion recovery—and (C) sagittal STIR—short TI inversion recovery—and corresponding axial and sagittal postcontrast images. Images (B) and (D) show significant resolution in the hyperintense signal in the area postrema with complete resolution of postcontrast enhancement in area postrema and cervicomedullary junction. Axial postcontrast images (E–H) show complete resolution of diffuse cortical and leptomeningeal enhancement in bilateral cerebral hemispheres.
Fig. 2 Follow-up magnetic resonance imaging. (A) Axial FLAIR—fluid attenuated inversion recovery—and (C) sagittal STIR—short TI inversion recovery—and corresponding axial and sagittal postcontrast images. Images (B) and (D) show significant resolution in the hyperintense signal in the area postrema with complete resolution of postcontrast enhancement in area postrema and cervicomedullary junction. Axial postcontrast images (E–H) show complete resolution of diffuse cortical and leptomeningeal enhancement in bilateral cerebral hemispheres.
Discussion
NMOSDs usually present with clinical manifestations corresponding to the involvement of long segments of spinal cord and optic nerves. Various less-frequent symptoms described include hypersomnolence, encephalopathy, and tonic spasms. Intractable nausea, vomiting, and hiccups can occur as part of area postrema syndrome.2
Initial reports and diagnostic criteria considered abnormal brain findings are unlikely in NMOSD. The detection of highly specific aquaporin 4 antibody (Nmo-IgG) and improvement in the knowledge gaps led to the comprehensive diagnostic criteria in which various other clinical and correlating imaging findings were considered compatible with the diagnosis of NMOSD. These regions include area postrema, dorsal medulla, periaqueductal region, periependymal areas of fourth and third ventricles, hypothalamus, thalamus long corpus callosal lesions, and subcortical deep white matter. However, cerebral cortex involvement was thought to be a feature against this diagnosis.3 Typical imaging findings of spinal cord involvement include long-segment signal change affecting more than three contiguous vertebral segments, central cord lesions, T1 hypointensities, and postcontrast enhancement. Optic nerve abnormalities include more than half the length involvement and posterior lesions close to chiasma.4
Area postrema syndrome is a well-defined entity in NMOSD. Clinical features include intractable nausea, vomiting, and hiccups lasting for more than 48 hours, and not responding to antiemetics and symptomatic medication. It can precede the typical optic neuritis or longitudinal extensive myelitis by months to years.5 Likely pathogenic mechanism is high expression of aquaporin 4 in area postrema. Other areas with high aquaporin 4 expression include diencephalon, periventricular and periependymal regions, and astrocyte foot processes, making these sites more vulnerable to immune-mediated inflammatory damage.6
Our patient also had clinical and imaging features compatible with area postrema syndrome, warranting further evaluation for NMOSD. Based on the significant abnormal MRI brain findings, ADEM was considered as a close differential diagnosis.
In one of the largest series on NMOSD, Kim et al observed abnormal MRI brain findings in nearly 75% but cerebral cortex involvement in just over 3% of the cohort, highlighting its rarity.7 Majority of the patients with cortex involvement were young females. Common clinical features were headache, confusion, seizure, and focal deficits. Parasagittal, frontal, parietal, and occipital lobes are common sites involved. In approximately half of the patients, cortical involvement was observed during initial presentation like in our present report. Coexisting leptomeningeal inflammation is common and cortex involvement is likely due to the leakage of inflammation from adjacent meningeal involvement and disruption of blood brain barrier.7 8
In NMOSD, vision impairment is generally attributed to the optic nerve or chiasma involvement. Homonymous hemianopia with unilateral occipital involvement was described in anti-MOG antibody positive NMOSD.9
Lalji et al described a very rare manifestation of cortical blindness as the exclusive cause of vision loss in aquaporin 4 antibody positive Sjogren syndrome associated NMOSD.10 In our patient also, absence of relative afferent pupillary defect, normally reacting pupils to light, normal fundus examination, presence of encephalopathy, and correlating occipital significant imaging abnormalities make the possibility of cortical blindness more likely cause of impaired vision than optic neuritis.
Accurate diagnosis is important as the treatment protocols in NMOSD are quite different from other demyelinating disorders like multiple sclerosis and ADEM.
Treatment modalities during acute attacks of NMOSD include intravenous methyl prednisolone 1 g daily for 3 to 5 days, continuation of oral corticosteroids with gradual dose reduction, and plasmapheresis in severe cases with poor response to steroids. Various steroid-sparing agents used for relapse prevention include azathioprine, methotrexate, mycophenolate, and rituximab.11
Conclusion
NMOSDs can have various atypical presentations. Although extensive cerebral cortical involvement is extremely uncommon in NMOSD, the presence of additional clinical manifestations and supportive imaging findings should alert the clinician for the prompt evaluation. Rarely, cortical blindness could be the underlying pathogenic mechanism for the vision impairment with or without the coexisting and more common optic neuritis. High index of clinical suspicion helps in the timely diagnosis and appropriate management of such atypical cases.
Conflict of Interest
None declared.
References
- Area postrema syndrome: frequency, criteria, and severity in AQP4-IgG-positive NMOSD. Neurology. 2018;91(17):e1642-e1651.
- [Google Scholar]
- International Panel for NMO Diagnosis. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. 2015;85(2):177-189.
- [Google Scholar]
- MRI characteristics of neuromyelitis optica spectrum disorder: an international update. Neurology. 2015;84(11):1165-1173.
- [Google Scholar]
- Intractable vomiting and hiccups as the presenting symptom of neuromyelitis optica. Ann Indian Acad Neurol. 2014;17(1):117-119.
- [Google Scholar]
- Neuromyelitis optica brain lesions localized at sites of high aquaporin 4 expression. Arch Neurol. 2006;63(7):964-968.
- [Google Scholar]
- cerebral cortex involvement in neuromyelitis optica spectrum disorder. J Clin Neurol. 2016;12(2):188-193.
- [Google Scholar]
- Cerebral cortex impairment in neuromyelitis optica spectrum disorder: a case report and literature review. Mult Scler Relat Disord. 2019;32:9-12.
- [Google Scholar]
- Anti-myelin oligodendrocyte glycoprotein antibody associated neuromyelitis optica spectrum disorder presenting homonymous hemianopia. Neurol Asia. 2019;24(4):373-375.
- [Google Scholar]
- Cortical blindness and not optic neuritis as a cause of vision loss in a Sjögren’s syndrome (SS) patient with the neuromyelitis optica spectrum disorder (NMOSD): challenges of ascribing demyelinating syndromes to SS: a case report. Medicine (Baltimore). 2017;96(34):e7454.
- [Google Scholar]
- Diagnosis and treatment of NMO spectrum disorder and MOG-encephalomyelitis. Front Neurol. 2018;9:888.
- [Google Scholar]




