
Translate this page into:
Diagnosing Muscular Dystrophies: Comparison of Techniques and Their Cost Effectiveness: A Multi-institutional Study
Kiran Preet Malhotra, MD, DNB, PDCC Department of Pathology, Dr. Ram Manohar Lohia Institute of Medical Sciences Gomti Nagar, Lucknow 226016, Uttar Pradesh India drkiranpreetmalhotra@yahoo.co.in
This article was originally published by Thieme Medical and Scientific Publishers Private Ltd. and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Abstract
Background The diagnosis of muscular dystrophies involves clinical discretion substantiated by dystrophic changes on muscle biopsy. The different subtypes of muscular dystrophy can be diagnosed using techniques to identify the loss of protein or molecular alterations.
Materials and Methods Clinically suspicious cases confirmed to have muscular dystrophy on muscle biopsy seen at two tertiary care centers in North India were enrolled for the study. Immunohistochemistry (IHC) for dystrophin, merosin, sarcoglycan, emerin, and dysferlin proteins was performed. The spectrum of muscular dystrophies diagnosed was analyzed. Cost of diagnosing the cases using IHC was estimated and compared with that of standard molecular tests available for the diagnosis of muscular dystrophies.
Statistics Descriptive statistics were used for data analysis. Mean and standard deviations were used for continuous variables, whereas categorical variables were analyzed using frequency percentage.
Results A total of 47 cases of muscular dystrophies were studied. This included nine cases of Duchenne, three cases of Becker’s dystrophy, and one dystrophinopathy carrier. One case of α, seven cases of β, and two cases of δ sarcoglycanopathy, along with two cases of facioscapulohumeral dystrophy and a single case of dysferlinopathy were detected. Genetic studies were required for a subset of 16 cases. The cost of using muscle biopsy and IHC was substantially lower than that of molecular methods for the identification of muscular dystrophy subtypes.
Conclusion We detailed an algorithmic approach for diagnosing muscular dystrophies using muscle biopsy. The prevalence of biopsy proven muscular dystrophies from two tertiary care centers in North India is compared with that from other centers. Genetic studies are currently of limited availability in India and are more expensive as compared with biopsy and IHC. Using these methodologies sequentially with a “biopsy first approach” may be the prudent approach for low-income countries.
Keywords
cost effectiveness
immunohistochemistry
muscular dystrophy
next generation sequencing
polymerase chain reaction

Introduction
Muscular dystrophies (MD) are genetically determined myopathies associated with histologic evidence of dystrophic changes seen on a muscle biopsy.1 Many of the proteins affected in muscular dystrophies can be localized to the sarcolemma, and their complete or partial deficiency is diagnostic. This can be identified using immunohistochemistry (IHC) which can be performed on frozen muscle biopsy tissue and permits rapid diagnosis.2 3 4 5 6 7
Immunohistochemical antibodies for diagnosing presence or absence of proteins including dystrophin, the sarcoglycan complex (α, β, γ, and δ), emerin, dysferlin, and merosin (laminin α 2) are currently available for use in muscle biopsy tissue. This allows the potential specific diagnosis of Duchenne and Becker MD, sarcoglycanopathies, Emery–Dreifuss MD, dysferlinopathy, miyoshi myopathy, and congenital MD.2 Loss of calpain-3 antibody (for diagnosing LGMD2A) can be currently detected by western blotting, not by IHC.8 However, for certain LGMDs like LGMD2L characterized by mutations in ANO5 gene, antibody testing is not available and they can currently be detected by mutation analysis alone.8
Routine histopathologic examination of muscle biopsy provides information about the architecture and can reveal dystrophic changes in muscle biopsies; however, the type of muscular dystrophies cannot be categorized on routine histopathology alone. IHC is helpful in identifying the presence or lack of specific proteins and can categorize the vast group of muscular dystrophies. Genetic studies show higher sensitivity for variation in protein expression but are technically difficult and expensive. Moreover, genetic studies can be used to assess dystrophinopathies alone and are not widely available in nonresearch settings for other muscular dystrophies. Second, numerous mutations exist which may cause loss of a single muscle protein; hence, it is not always feasible to perform genetic analysis for multiple mutations in these cases as it is labor intensive and time consuming.2
The full-length dystrophin protein is a high molecular mass protein with four main domains.2 Among mutations in the dystrophin gene, about two-thirds are deletions, 5 to 10% are duplications and the remainder are point mutations. These point mutations are difficult to identify with standard polymerase chain reaction but IHC easily identifies all mutations that lead to the formation of a stop codon and hence absence of protein.2 Among molecular tests available for muscular dystrophy diagnosis are multiplex ligation probe amplification (MLPA), array-based comparative genomic hybridization (CGH) and next generation sequencing (NGS, Table 1).9 10 11 12 13 14 15 16 17 18 19 20 21
|
IHC analysis |
Western blotting |
Multiplex PCR |
MLPA |
cGH |
NGS |
|
|---|---|---|---|---|---|---|
|
Abbreviations: AR, autosomal recessive; cGH, comparative genomic hybridization; FKRP, fukutin-related protein; IHC, immunohistochemistry; LGMD, limb-girdle muscular dystrophy; MLPA, multiplex ligation probe amplification; NGS, next generation sequencing; PCR, polymerase chain reaction. |
||||||
|
Sample requirements |
Fresh frozen muscle biopsy tissue |
Fresh frozen muscle biopsy tissue |
Whole blood |
Whole blood, chorionic villi, and muscle biopsy |
Whole blood |
Whole blood |
|
Procedure specifications |
Muscle biopsy procedure comparatively expensive and invasive may require general anesthesia in children |
Muscle biopsy procedure comparatively expensive, invasive, may require general anesthesia in children |
Inexpensive blood sample collection |
Inexpensive blood sample collection |
Inexpensive blood sample collection |
Inexpensive blood sample collection |
|
Availability of test |
Performed in specialized laboratories |
Performed in specialized laboratories |
Performed in specialized laboratories |
Performed in specialized laboratories |
Performed in specialized laboratories |
Performed in high end research settings |
|
Time required |
About six person hours |
Overnight |
Overnight |
2 days |
4 days |
Hours to days (depending upon platform used) |
|
Instrumentation required |
Manual or automated IHC autostainer |
SDS-polyacrylamide gel electrophoresis system and Densitometry |
PCR analyzers |
PCR followed by genetic analysis |
Oligonucleotide probe hybridization, image scanning, and software analysis |
Next generation sequencer. Available sparingly in high end laboratories. |
|
Standardization of procedure |
Easy |
Easy |
Easy |
Difficult Requires expertise in data analysis |
Difficult |
Difficult. Requires expertise in data analysis. |
|
Current availability for |
Dystrophins, sarcoglycans, dysferlin, FKRP, merosin, emerin. Calpain and caveolin cannot be diagnosed. No antibody available for LGMD 2L with mutation in ANO5 |
Calpain, Caveolin can be diagnosed in addition to those diagnosable by IHC |
Dystrophinopathies and sarcoglycanopathies |
Dystrophinopathies and sarcoglycanopathies |
Dystrophinopathies and sarcoglycanopathies |
Dystrophins mostly. Mutation testing is currently being tested for many LGMDs |
|
Cost of test |
Low |
Moderate |
Moderate |
Moderate (similar to PCR) |
High |
High |
|
Parameter tested |
Final protein product |
Final protein product |
Gene deletions |
Gene deletions or duplications excluding intronic rearrangements. Point mutations not detected. |
Gene deletions or duplications including intronic rearrangements. Point mutations not detected |
Gene deletions, duplications and point mutations |
|
Sensitivity |
High. Tests protein presence or absence. Example total three proteins for dystrophin abnormalities. Detects almost 100% of dystrophinopathies. |
High. Tests protein presence or absence. Example total three proteins for dystrophin abnormalities. Detects almost 100% of dystrophinopathies. Detects LGMD 2A with approximately 53% sensitivity and 85% specificity. |
Detects almost 68.7% of dystrophinopathies |
Detects almost 75% of dystrophinopathies |
High. Tests each gene locus |
High. Can detect abnormality in entire genome |
|
Usability of test |
Poor in advanced disease with fatty replacement of muscle. Useful in AR LGMD with loss of function; alteration or loss of one mutant allele is difficult to detect in AD forms. Secondary reduction of expression of associated proteins in LGMD requires caution in interpretation |
Cannot differentiate small amounts of residual protein from revertant fiber protein expression. Secondary reduction of expression of associated proteins in LGMD requires caution in interpretation |
Gene duplications not detected |
About one third of MLPA negative cases had dystrophinopathy on muscle IHC |
Limit to the number of genes that can be tested simultaneously |
Improves with availability of muscle biopsy protein analysis |
|
Prognostic value |
Amount of membrane protein correlates better with patient phenotype |
Amount of membrane protein correlates better with patient phenotype |
Does not predict phenotypic manifestation |
Does not predict phenotypic manifestation |
Does not predict phenotypic manifestation |
Does not predict phenotypic manifestation |
|
Use in therapeutic trials |
Good for assessing outcome of therapeutic trials for restoring absent membrane proteins |
Good for assessing outcome of therapeutic trials for restoring absent membrane proteins |
Cannot predict phenotypic outcome; less likely to aid therapeutic trials |
Cannot predict phenotypic outcome; less likely to aid therapeutic trials |
Cannot predict phenotypic outcome; less likely to aid therapeutic trials |
Cannot predict phenotypic outcome; less likely to aid therapeutic trials |
Objectives
The current study aimed to assess the profile of muscular dystrophies, characterize them using minimalistic immunohistochemical panels, and evaluate the cost effectiveness of IHC as compared with genetic studies in the diagnosis and categorization of muscular dystrophies in a resource-limited setting.
Materials and Methods
The present study was performed at two tertiary care referral hospitals: King George Medical University, Lucknow (KGMU) and Dr. Ram Manohar Lohia Institute of Medical Sciences, Lucknow (RMLIMS). Consecutive clinically suspected cases of muscular dystrophies presenting at the departments of neurology at KGMU and RMLIMS were recruited over a period of 2 years. The biopsy analysis was performed at the department of pathology, RMLIMS.
-
Clinical assessment: Detailed history included the following factors: age, sex, family history, age of onset, topography of muscle involvement, and presence of calf hypertrophy. Physical examination as well as manual muscle testing utilizing the Standard Medical Research Council grading system for the assessment of power of muscle and neuroelectrophysiology was done in each case.22
-
Laboratory investigations: Routine hematological tests, serum calcium and creatine phosphokinase levels were evaluated in all cases.
-
Muscle biopsy processing: Cases suspected to have muscular dystrophy based on history, clinical, and serological profile underwent muscle biopsy after informed consent. Patients with bleeding disorders or on anticoagulants, those with a recent history of muscle infection or trauma to the biopsy site and those with myopathies other than muscular dystrophy like inflammatory, hypothyroid, or osteomalacic myopathy were excluded from the study. A muscle showing moderate affliction was selected for biopsy. Open muscle biopsy was performed, and tissue transported without delay to the laboratory in a fresh state wrapped in saline moistened gauze, without any fixative. The tissue was divided into two equal portions. One portion was snap frozen in isopentane precooled in liquid nitrogen and serially sectioned in the cryostat. Sections were used for hematoxylin and eosin and IHC staining. The second portion was routinely processed in formalin and paraffin embedded.
-
Immunohistochemical studies: These were performed on frozen muscle tissue using monoclonal antibodies to dystrophin N domain (1:20), dystrophin C domain (1:20), and dystrophin rod domain (1:5); sarcoglycans α, β, γ, and δ (all at a dilution of 1:20); dysferlin (1:25) and Merosin (1:50). All primary antibodies procured from Novocastra, United Kingdom. Frozen sections were hydrated with Tris-buffered saline and incubated in endogenous blocking solution for 30 minutes, followed by washing and incubation in primary antibody for 2 hours. Treatment with HRP-tagged secondary antibody (Envision system from Dako; Glosstrup, Denmark) was done for 30 minutes. Diaminobenzidine was used as substrate chromogen.
-
Biopsy assessment: Morphologic features were evaluated using Hematoxylin and Eosin stained sections. Endomysial fibrosis was assessed using Masson Trichrome staining. Biopsies suggestive of muscular dystrophy on histomorphology were further categorized using IHC.
-
IHC assessment: IHC panels were tailored according to the history and clinical profile of individual cases. Biopsies showing fixation artifacts or poor tissue preservation on H and E sections of frozen tissue were excluded from further IHC workup. Merosin staining was performed in all cases to ensure integrity of sarcolemmal membrane. Cases showing loss of merosin staining were excluded from further IHC evaluation. Parallel positive controls (from biopsies of known cases of inflammatory myopathies) were run in each case.
-
Cost analysis: This was performed for the diagnostic panel used in our cases. Cost of biopsy procedure, and IHC was calculated for cases based on the effective cost at our Institute. For cost comparison, a representative cohort was used with the assumption that genetic and molecular testing were performed on blood samples alone and muscle biopsy was avoided. Standard cost of molecular and genetic tests in reputed laboratories was used for the purpose of comparison.
Statistical Analysis
Descriptive statistics were used for data analysis. Mean and standard deviation were used for continuous variables, whereas categorical variables were analyzed using frequency percentage. A p-value was calculated for the association of clinical and histological findings with the final diagnosis.
Results
A total of 47 muscle biopsies from suspected cases of muscular dystrophies were received. Histomorphology of all biopsies showed features of dystrophy which included presence of fibrosis in the endomysium, rounded fibers with variable muscle fiber size, numerous internal nuclei, and presence of degenerating and regenerating fibers. Few cases showed split fibers and endomysial inflammation in addition. Of the 47 cases, five cases showed poor tissue preservation on H and E sections, and were excluded from further IHC analysis. Results of IHC analysis on muscle biopsies of these patients, and their clinical features are presented in Table 2. The clinical, electrophysiological, and biochemical features of the cases are presented in Table 3.
|
Diagnosis |
Number of cases |
Mean age ±SD (y) at presentation |
Male: Female Ratio |
Positive Family History (number of cases) |
|---|---|---|---|---|
|
Abbreviations: DMD, Duchenne muscular dystrophy; FSHD, fascioscapulohumeral dystrophy; SD, standard deviation. |
||||
|
Dystrophinopathy |
13 |
9.92 ± 2.43 |
12:1 |
2 |
|
DMD |
9 |
|||
|
Becker’s dystrophy |
3 |
|||
|
Dystrophinopathy carrier |
1 |
|||
|
Sarcoglycanopathy |
10 |
20.6 ± 10.09 |
9:1 |
2 |
|
Sarcoglycan α |
1 |
|||
|
Sarcoglycan β |
7 |
|||
|
Sarcoglycan γ |
0 |
|||
|
Sarcoglycan δ |
2 |
|||
|
Dysferlinopathy |
1 |
24 |
1:0 |
0 |
|
MD not further categorized |
18 |
|||
|
MD Dystrophinopathy & sarcoglycanopathy excluded |
16 |
24 ± 13.5 |
1.2:1 |
2 |
|
MD FSHD |
2 |
25 ± 15.5 |
1:1 |
0 |
|
Total cases analyzed |
42 |
18.8 |
5:1 |
6 |
|
S No. |
Type of muscular dystrophy |
Lower limb weakness |
Upper limb weakness |
Facial weakness |
Calf hypertrophy |
Myopathic pattern on electromyography |
Nerve conduction studies |
Mean serum CPK (U/L) |
Serum calcium (g/dL) |
|---|---|---|---|---|---|---|---|---|---|
|
Abbreviations: CPK, creatine phosphokinase; FSHD, fascioscapulohumeral dystrophy. |
|||||||||
|
1. |
Dystrophinopathy |
13 |
7 |
0 |
6 |
11 |
Normal |
4,904 ± 3,342 |
9.7 |
|
2. |
Sarcoglycanopathy |
10 |
5 |
0 |
3 |
8 |
Normal |
4,264 ± 2,023 |
9.6 |
|
3. |
Dysferlinopathy |
1 |
1 |
0 |
0 |
1 |
Normal |
3,456 |
9.4 |
|
4. |
Muscular dystrophy not further categorized |
||||||||
|
MD dystrophinopathy and sarcoglycanopathy excluded |
14 |
12 |
0 |
1 |
13 |
Normal |
3,174 ± 2,446 |
9.2 |
|
|
MD FSHD |
0 |
2 |
2 |
0 |
1 |
Normal |
3,193 ± 3,498 |
9.3 |
|
|
5. |
Total cases analyzed |
38 |
27 |
2 |
10 |
34 |
4,642 average |
9.4 average |
|
Dystrophinopathies
A total of 13 cases showed absence or diminution of sarcolemmal dystrophin staining and were grouped as dystrophinopathies. Of these, nine showed complete absence of N, C, and Rod domain staining, and were consistent with a diagnosis of Duchenne muscular dystrophy (DMD). Along with dystrophins mild secondary reduction of sarcoglycans α and β was also seen in one of the DMD cases. Three cases showed weak but retained membrane staining for one or more of the three dystrophin antibodies, and were consistent with Becker’s muscular dystrophy. A single female patient showed reduced staining for dystrophin panel and was diagnosed as DMD carrier.
Sarcoglycanopathies
Ten cases showed abnormal sarcoglycan staining and grouped as sarcoglycanopathies. Seven cases were categorized as β sarcoglycanopathy of which four showed complete absence, while three showed reduced immunostaining of β sarcoglycan. Secondary mild reduction of α sarcoglycan staining was also seen in four cases diagnosed as β sarcoglycanopathy. A single case of α sarcoglycanopathy was identified, which showed absence of α sarcoglycan staining with secondary mild reduction of β sarcoglycan. Two cases diagnosed as δ sarcoglycanopathy showed complete absence of δ sarcoglycan; of these, one case also showed secondary reduction of the other three sarcoglycans.
Dysferlinopathy
With complete absence of dysferlin, one case was diagnosed as having dysferlinopathy, staining for sarcoglycans and dystrophins being normal.
Muscular Dystrophy not further Categorized
A total of 18 cases showed no change in immunoreactivity in any of the IHC panels used. Among these, two cases were clinically suspected to be Fascioscapulohumeral dystrophy (FSHD) based on the pattern of muscle weakness. These were diagnosed as FSHD on the basis of histopathologic findings and exclusion of IHC abnormalities. Out of the remaining 16 cases, 14 showed presence of weakness in all four limbs and two showed presence of upper limb weakness alone. These cases could only be diagnosed as muscular dystrophy on histomorphology and could not be categorized further immunophenotypically.
The results obtained using our diagnostic approach to muscular dystrophies using IHC panels is presented in Fig. 1.
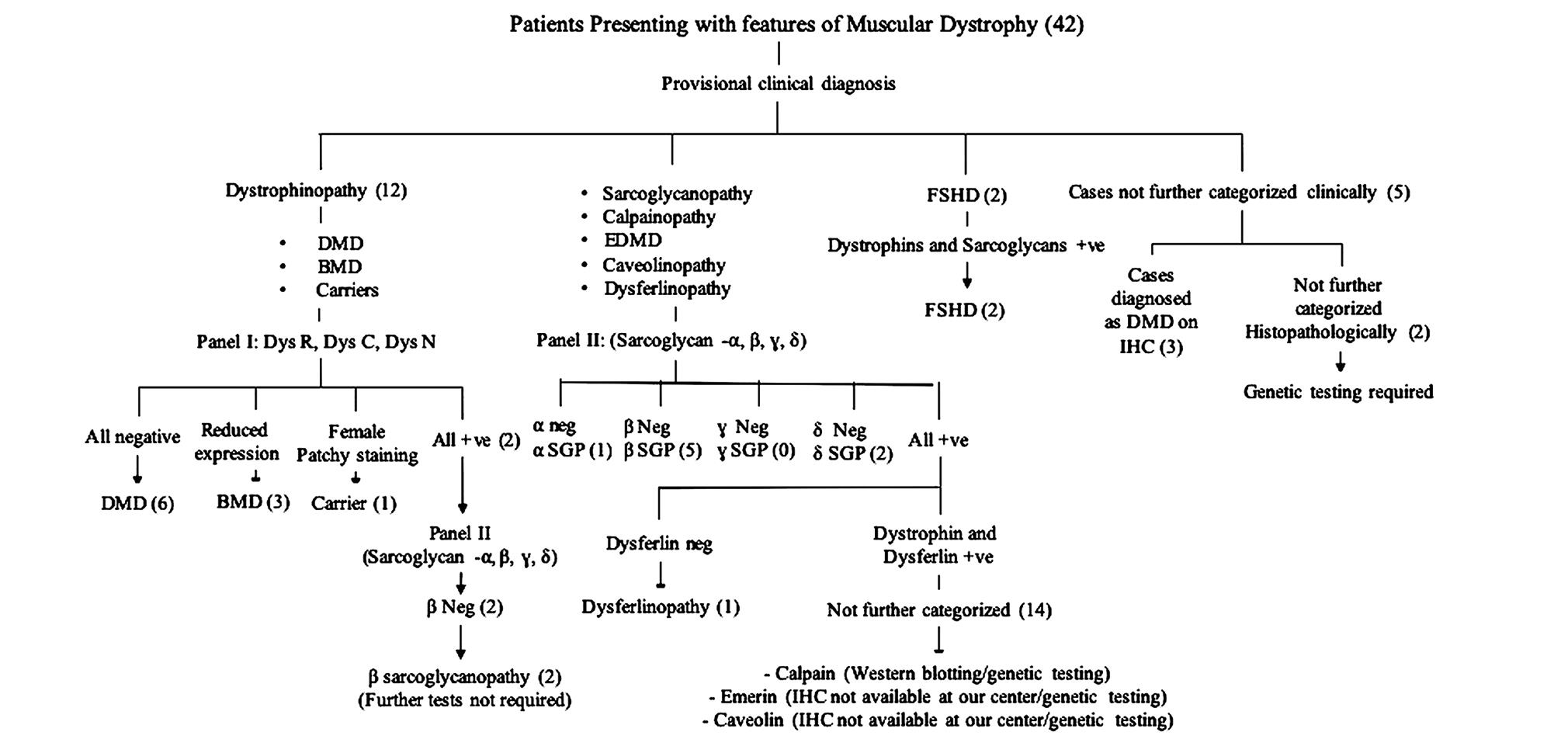
-
Fig. 1 Patients presenting with features of muscular dystrophy (42). BMD, Becer’s muscular dystrophy; DMD, Duchenne muscular dystrophy; EDMD, emery dreifuss muscular dystrophy; FSHD: fasciosc apulohumeral muscular dystrophy; SGP, sarcolgycanopathy.
Fig. 1 Patients presenting with features of muscular dystrophy (42). BMD, Becer’s muscular dystrophy; DMD, Duchenne muscular dystrophy; EDMD, emery dreifuss muscular dystrophy; FSHD: fasciosc apulohumeral muscular dystrophy; SGP, sarcolgycanopathy.
Cost Minimization Analysis
An economic analysis was done in which we compared the net costs of determining the diagnosis and subtype of muscular dystrophies utilizing primarily muscle biopsy (with or without resort to subsequent genetic studies) versus using genetic analysis alone. The cost of methodologies of muscle biopsy (using IHC studies), and whole blood genetic analysis was compared to establish the cheapest alternative among them.
The minimum cost of diagnosing muscular dystrophies using a biopsy first approach (followed by use of genetic studies as required) was Rs 3,12,200, and minimum cost calculated for diagnosing all cases by genetic studies was Rs 5,79,600 which was significantly higher (Table 4).
|
S.No |
Diagnostic method used |
Cost per test in INR |
Number of cases tested |
Total cost in INR |
|---|---|---|---|---|
|
Abbreviations: cGH, comparative genomic hybridization; IHC, immunohistochemistry; INR, Indian rupee; MLPA, multiplex ligation probe amplification; PCR, polymerase chain reaction. |
||||
|
1 |
“Biopsy first” approach |
|||
|
1a |
Cost of biopsy procedure |
300 |
42 |
12,600 |
|
1b |
Histology and immunohistochemistry studies |
1,400 |
42 |
58,800 |
|
1c |
Cost of genetic studies for cases left undiagnosed by histopathology and IHC |
16 |
||
|
1c(i) |
Multiplex DNA PCR with Begg’s and Chamberlain’s primer |
3,800 |
16 |
60,800 |
|
1c(ii) |
Comparative genomic hybridization |
1,05,000 |
16 |
16,80,000 |
|
1c(iii) |
Multiplex ligation probe amplification |
12,000 |
16 |
1,92,000 |
|
1d |
Cost of next generation sequencing for remaining 1/3 cases not diagnosed by above methods |
30,000 |
6 |
1,80,000 |
|
Total cost range (depending upon type of genetic study used) 1a+1b+1c (either of i, ii, or iii) + 1d |
42 |
3,12,200 (i); 19,31,400 (ii); 4,43,400 (iii) |
||
|
2 a |
“Nonbiopsy/ genetic analysis approach” |
42 |
||
|
2b (i) |
Using next generation sequencing alone |
30,000 |
42 |
12,60,000 |
|
2 b (ii) |
Multiplex DNA PCR with Begg’s and Chamberlain’s primer studies |
3800 |
42 |
1,59,600 |
|
2 b (iii) |
Comparative genomic hybridization |
1,05,000 |
42 |
44,10,000 |
|
2b (iv) |
Multiplex ligation probe amplification |
12,000 |
42 |
5,04,000 |
|
2c |
Cost of next generation sequencing for remaining 1/3 cases not diagnosed by multiplex PCR/cGH/MLPA |
30,000 |
14 |
4,20,000 |
|
Total cost for nonbiopsy/genetic studies approach (depending upon type of genetic study used) 2b (either of i, ii, iii or iv) + 2c |
42 |
12,60,000 (i); 5,79,600(ii); 48,30,000 (iii); 9,24,000 (iv) |
||
|
Incremental cost of genetic studies over the biopsy first approach 5,79,600–3,12,200 |
42 |
2,67,400 |
||
|
Minimal cost per patient diagnosed using biopsy first approach |
Per patient |
7433 |
||
|
Minimal cost per patient diagnosed using nonbiopsy/genetic studies approach |
Per patient |
13,800 |
||
Discussion
The current study analyzed 47 consecutive patients from all age groups, clinically diagnosed to have muscular dystrophy. The clinical, biochemical, muscle biopsy histopathology, and IHC profile of all cases was studied. A minimalistic IHC panel was tailored for each case based on the clinical history, examination, and histomorphology. The cost of biopsy first approach using IHC studies primarily was compared with cost required to diagnose and categorize muscular dystrophies using molecular tests, and the former method proved more cost effective.
Total 31% of all cases were categorized as dystrophinopathies, including DMD (69.2%), BMD (23%), and one female dystrophinopathy carrier. Of the 10 cases of sarcoglycanopathies (23% of total), 70% showed β, 10% α, and 20% δ sarcoglycanopathy; a solitary case of dysferlinopathy (2.3%) was diagnosed along with two cases of facioscapulohumeral dystrophy (4.7%). No case of Emery–Dreifuss disease was identified. Total 16 cases (38.1%) showing clinical and histologic evidence of muscular dystrophy could not be further categorized due to absence of immunohistochemical alterations in any of the IHC panels used.
Majority of dystrophinopathies were detected in children less than 10 years of age (mean age = 9.9 years). Similarly, in a study from Gujarat, India, the mean age of DMD patients was 10.75 years at presentation.23 Most patients were males, relating to the X-linked inheritance. One female patient was diagnosed as a DMD carrier. Patients with sarcoglycanopathies had a mean age of 11.8 years at disease onset and 20.6 years at disease diagnosis. In a report from Northern India, mean age of sarcoglycanopathy patients was 7.2 years with age range of 5 to 20 years at onset of symptoms.7 In another Indian study by Khadilkar et al, patients with sarcoglycanopathy had a mean age of 25.84 years (with age range of 8–42 years) and age at onset of symptoms was 15 years (with age range of 5–37 years).4 In our series, cases with sarcoglycanopathy had a high male:female ratio of 9:1. Other series on sarcoglycanopathy have reported male: female ratios of 1.1:1 and 1.5:1.6 7 One male patient with dysferlinopathy and two cases of FSHD (one male and one female) were also diagnosed in our series. Positive family history could be obtained only in six cases (14.3%). Similarly, in a study on 106 dystrophinopathy patients, only 17% had a positive family history.24
The commonest presenting symptom was proximal symmetrical lower limb weakness (in 38 of 42 patients). Distal weakness was found in three cases, one of DMD and two which could not be categorized further. Ten patients presented with calf hypertrophy, including 46% cases of dystrophinopathy and 37.15% cases of sarcoglycanopathy. Rao et al found calf hypertrophy in 87% of dystrophinopathy cases.23 A high proportion of sarcoglycanopathy cases also showed presence of calf hypertrophy including 69% cases reported by Sharma et al and 44% reported by Khalidar et al.4 7 Scapular winging was seen in four of our cases; among them two were FSHD, one DMD and two dystrophy cases which could not be categorized further.
In our study, the mean CK value was 4642.3 IU/L. In dystrophinopathy group, it was 4,904 IU/L (mean CPK of 5,568 IU/L in DMD and 4,486 IU/L in BMD patients). Similar elevated CK values have been reported in dystrophinopathies reported from Gujarat (mean CPK 7218.4 in DMD and 5,574 IU/L in BMD).23 The sarcoglycanopathy patients showed a mean CPK of 4,264 IU/L in our series, whereas it was 7,853 IU/L in a series by Nalini et al.6 The nerve conduction velocity was normal in all patients. Electromyography (EMG) pattern was myopathic in 80.9% cases among which dsytrophinopathy cases showed myopathic pattern in 84% cases and sarcoglcanopathy in 80% cases. One of the two FSHD cases and the dysferlinopathy case also showed myopathic pattern. In a study by Nalini et al, 65.1% of sarcoglycanopathy cases showed myopathic pattern on EMG.6
The MDs were diagnosed by the presence of “dystrophic” changes on muscle biopsy and further categorized using IHC on fresh frozen muscle biopsy tissue. In the present study, nine cases showed complete absence of dystrophin rod domain, C-terminus domain, and N-terminus domain (except for few revertant fibers); and were categorized as DMD. Incomplete patchy sarcolemmal immunostaining of dystrophin antibodies was seen in the three cases of BMD. Barresi et al also recommend the use of multiple antibodies focused at numerous sites of the large dystrophin molecule to prevent false negative results which may arise due to loss of epitopes away from the site recognized by a particular antibody. A panel of antibodies focused against N-terminal, C terminal or rod domain is hence routinely used for IHC.25 A female carrier was also detected in our series based on patchy expression of dystrophin antibodies in the muscle biopsy. Among 106 suspected dystrophinopathy cases, Freund et al found immunohistochemical abnormalities on muscle biopsy in four females (one DMD patient and three carriers); however, none of these four could be detected using PCR on peripheral blood leucocytes.24 MLPA has also been reported to be of use in detecting female carriers where the male proband harbors a deletion or duplication of dystrophin gene. For point mutations, direct sequencing is reportedly a better method than MLPA analysis.26
In the present study, we could classify 10 cases (21% of all MD cases) as sarcoglycanopathy. Seven cases of β SGP, two of δ SGP, one of α SGP, one dysferlinopathy, and two FSHD were diagnosed. The reported incidence of sarcoglycanopathies among LGMD cases in India varies from 11.8 to 46.2%.4 7 It may be noted that whether this actually represented the LGMD genotype as a whole is doubtful. The lack of use of a uniform definition for including patients, as well as referral bias, impart a significant amount of heterogeneity to studies whereby no study from the Indian subcontinent could find a place in the meta-analysis and systematic review focused at detailing epidemiologic data on muscular dystrophies.27
Few studies on sarcoglycanopathies confirmed on IHC or genetic analysis are reported from India. The commonest IHC pattern in sarcoglycanopathies was that of loss of multiple sarcoglycans, in several series including ours.4 7 Multiple losses were noted in 70% cases in our study and ranged from 38.5 to 84% in other Indian series.4 7 Intricate interaction among the sarcoglycan subunits and destabilization of the complex is probably the basis for this. Similar to our study where 20% cases showed isolated loss of β sarcoglycan, Nalini et al reported loss of β sarcoglycan alone in 19% cases and Khadilkar et al in 12%.4 6 Deficient β sarcoglycan expression was followed by that of δ sarcoglycan in our study, similar to the findings of Khadilkar et al.4 This is understandable since β and δ subunits form the core of the sarcoglycan complex. On the contrary, no case with isolated deficiency of β or δ sarcoglycan was noted among 13 cases reported from Delhi, where γ sarcoglycanopathy was followed in frequency by α sarcoglycanopathy.7 IHC may hence guide genetic analysis by either defining an isolated affected gene or highlighting the one most severely affected from amongst multiple affected ones.4 7
A single case of dysferlinopathy (3.5% of LGMD) and two of FSHD (7% of LGMD) were noted in our series, whereas they proportioned 10.2 and 28% respectively in a series by Sharma et al.7 Data on these entities are not available from other large series from India. A large proportion of LGMD cases remained unclassified on IHC, this included 55% cases from our series, matched closely by 54% reported by Khadilkar and 49% by Sharma et al.4 7 This group may comprise either patients who have missense mutations in a SG gene (without change in protein expression) or nonsarcoglycan deficient forms of LGMD. Defects in several nonsarcoglycan genes presenting as LGMDs have been reported and include calpain, titin, dystroglycans, fukutin, and anoctamin 5 among others, which were not analyzed in the present study.28 29 IHC analysis is not available for use in muscle biopsy tissue for most of these and molecular analysis would be essential to ascertain the primary abnormality. However, currently IHC and western blotting are considered the gold standards for diagnosis of dysferlin, sarcoglycans, caveolin 3, and telethonin antibodies.29
Though data on calpainopathy is limited from India, a recent Indian study using immunoblotting identified 47% of all LGMD cases to be due to calpainopathy; this may mean that calpainopathy is the commonest form of LGMD in India, similar to recent epidemiologic data from across the globe.30 Immunostaining prior to molecular analysis can identify approximately half of the protein abnormalities in cases presenting with LGMD, thereby allowing genetic analysis to focus on required genes. While supporting our findings, Nalini et al in their study also suggest that genetic testing is necessary for prenatal diagnosis of muscular dystrophies and genetic counselling.6
We also analyzed the cost of using IHC as a primary specific diagnostic tool versus direct genetic diagnostic approach (Table 4). It is clear from our analysis that the biopsy first approach was more economic than genetic studies.
There were some limitations of the present study. Total 47 cases were included in our study, but five cases could not be analyzed due to poor preservation of the tissue. In some cases, biopsy from severely affected muscle was sent which was completely replaced by fat and connective tissue, so IHC interpretation was not possible in those cases. We were unable to perform western blotting for detecting suspected calpainopathies due to unavailability of the antibody for our study. Though the cost of diagnosing muscular dystrophies using various methods was compared, the genetic analysis was not performed at our center and the applicable cost at other reputed centers in India was used for comparison. Such cost comparison performed on a larger group of patients would probably provide a more holistic cost benefit ratio.
Conclusion
In conclusion, our study provided data on biopsy proven muscular dystrophies from two tertiary care centers in North India and compared their prevalence with data from other centers in India. Biopsy studies can be utilized for definite diagnosis of several muscular dystrophies and also narrow the differential diagnosis so that appropriate genetic studies can be planned. Genetic studies are currently of limited availability in India and are more expensive as compared with biopsy and IHC. Currently, no specific therapies exist for these muscular dystrophies. However, correct identification of the subtype aids prediction of subsequent phenotype and behavior of the disorder. Genetic and familial counseling can also be specifically directed. Moreover, targeted therapy may be available in the future with advancement in knowledge regarding these molecules. Using these methodologies sequentially with a “biopsy first approach” may be the prudent approach for low income countries.
Conflict of Interest
None declared.
References
- Recent advances in diagnosis of the childhood muscular dystrophies. J Paediatr Child Health. 1997;33(3):195-201.
- [Google Scholar]
- Muscle biopsy: A Practical Approach. Philadelphia, USA: Elsevier, Saunders; 2007. In: eds.
- [Google Scholar]
- Muscle Disease: Pathology and Genetics. (2nd ed.). Chichester, UK: Wiley Blackwell; 2013.
- [Google Scholar]
- Sarcoglycanopathies: a clinico-pathological study. Neurol India. 2007;55(2):117-121.
- [Google Scholar]
- Sarcoglycanopathy: clinical and histochemical characteristics in 66 patients. Neurol India. 2010;58(5):691-696.
- [Google Scholar]
- Sarcoglycanopathies: a clinicopathological study of 13 cases [corrected] Neurol India. 2004;52:446-449.
- [Google Scholar]
- A practical approach to molecular diagnostic testing in neuromuscular diseases. Phys Med Rehabil Clin N Am. 2012;23(3):589-608.
- [Google Scholar]
- Use of capillary Western immunoassay (Wes) for quantification of dystrophin levels in skeletal muscle of healthy controls and individuals with Becker and Duchenne muscular dystrophy. PLoS One. 2018;13(4):e0195850.
- [Google Scholar]
- Detection of 98% of DMD/BMD gene deletions by polymerase chain reaction. Hum Genet. 1990;86(1):45-48.
- [Google Scholar]
- A comprehensive genomic approach for neuromuscular diseases gives a high diagnostic yield. Ann Neurol. 2015;77(2):206-214.
- [Google Scholar]
- Small mutations detected by multiplex ligation-dependent probe amplification of the dystrophin gene. Genet Test Mol Biomarkers. 2009;13(3):427-431.
- [Google Scholar]
- Nalini A, Singh RJ, Gayathri N. A comparative study of mPCR, MLPA, and muscle biopsy results in a cohort of children with Duchenne muscular dystrophy: a first study. Neurol India. 2015;63(1):58-62.
- [Google Scholar]
- Detecting copy number variations in autosomal recessive limb-girdle muscular dystrophies using a multiplex ligation-dependent probe amplification (MLPA) assay. Mol Cell Probes. 2009;23(1):55-59.
- [Google Scholar]
- Molecular diagnosis of Duchenne/Becker muscular dystrophy: enhanced detection of dystrophin gene rearrangements by oligonucleotide array-comparative genomic hybridization. Hum Mutat. 2008;29(9):1100-1107.
- [Google Scholar]
- Motor chip: a comparative genomic hybridization microarray for copy-number mutations in 245 neuromuscular disorders. Clin Chem. 2011;57(11):1584-1596.
- [Google Scholar]
- Microarray-based mutation detection in the dystrophin gene. Hum Mutat. 2008;29(9):1091-1099.
- [Google Scholar]
- Whole dystrophin gene analysis by next-generation sequencing: a comprehensive genetic diagnosis of Duchenne and Becker muscular dystrophy. Mol Genet Genomics. 2014;289(5):1013-1021.
- [Google Scholar]
- Targeted next-generation sequencing as a comprehensive test for patients with and female carriers of DMD/BMD: a multi-population diagnostic study. Eur J Hum Genet. 2014;22(1):110-118.
- [Google Scholar]
- Genetic diagnosis of Duchenne/Becker muscular dystrophy using next-generation sequencing: validation analysis of DMD mutations. J Hum Genet. 2016;61(6):483-489.
- [Google Scholar]
- Genetic diagnosis of Duchenne and Becker muscular dystrophy using next-generation sequencing technology: comprehensive mutational search in a single platform. J Med Genet. 2011;48(11):731-736.
- [Google Scholar]
- Medical Research Council. Aids to examination of the peripheral nervous system. Memorandum no. 45. London: her majesty’s stationary office. 1976. Available at: https://mrc.ukri.org/documents/pdf/aids-to-the-examination-of-the-peripheral-nervous-system-mrc-memorandum-no-45-superseding-war-memorandum-no-7/. Accessed April 4, 2020
- Duchenne/Becker muscular dystrophy: a report on clinical, biochemical, and genetic study in Gujarat population, India. Ann Indian Acad Neurol. 2014;17(3):303-307.
- [Google Scholar]
- Duchenne and Becker muscular dystrophy: a molecular and immunohistochemical approach. Arq Neuropsiquiatr. 2007;65(1):73-76.
- [Google Scholar]
- From proteins to genes: immunoanalysis in the diagnosis of muscular dystrophies. Skelet Muscle. 2011;1(1):24.
- [Google Scholar]
- Carrier detection in Duchenne muscular dystrophy using molecular methods. Indian J Med Res. 2013;137(6):1102-1110.
- [Google Scholar]
- A systematic review and meta-analysis on the epidemiology of the muscular dystrophies. Can J Neurol Sci. 2016;43(1):163-177.
- [Google Scholar]
- Limb-girdle muscular dystrophies: where next after six decades from the first proposal (Review) Mol Med Rep. 2014;9(5):1515-1532.
- [Google Scholar]
- The 2014 version of the gene table of monogenic neuromuscular disorders (nuclear genome) Neuromuscul Disord. 2013;23(12):1081-1111.
- [Google Scholar]
- Limb girdle muscular dystrophy type 2A in India: a study based on semi-quantitative protein analysis, with clinical and histopathological correlation. Neurol India. 2010;58(4):549-554.
- [Google Scholar]




