
Translate this page into:
Developmental delay, seizures, and behavioral disorder caused by new SETD1B mutation: A case report
 , Yelena Shayakhmetova1, Neilya Akimzhanova2, Marina Grigolashvili1,
, Yelena Shayakhmetova1, Neilya Akimzhanova2, Marina Grigolashvili1,
*Corresponding author: Marina Grigolashvili, Department of Neurology, Psychiatry, and Rehabilitation, Karaganda Medical University, Karaganda, Kazakhstan. grigolashvili@qmu.kz
-
Received: ,
Accepted: ,
How to cite this article: Seryogina A, Mukusheva A, Shayakhmetova YV, Akimzhanova N, Grigolashvili M. Developmental delay, seizures, and behavioral disorder caused by new SETD1B mutation: A case report. J Neurosci Rural Pract. 2024;15:603-6. doi: 10.25259/JNRP_339_2024
Abstract
The article presents a clinical observation of a patient with atypical absences, myoclonia, psychomotor, and speech retardation. Whole-exome sequencing of the patient’s genetic material revealed a previously undescribed mutation in the SETD1B gene. The phenotypic picture of our case is similar to the data of patients with variants in the SETD1B gene, previously described in the scientific literature. The majority of the identified mutations occur in the catalytic domains, single-electron transfer (SET) and N-single-electron transfer (N-SET). There are only 6 cases in the RNA binding domain (RNA recognition motif), accounting for 3 exons of the gene. However, regardless of the location and type of mutation, the phenotypic manifestations of the described cases have common features. We want to draw the attention of the global scientific community for further research and a better understanding of the genotype-phenotypic correlation in SETD1B-associated nervous system disorder.
Keywords
SETD1B
Atypical absences
Epilepsy
Myoclonus
Developmental delay
Genetic disease
Whole-exome sequencing

INTRODUCTION
Mutation in the SETD1B gene is represented by developmental delay, autism spectrum disorder, and seizures. In most cases, psychomotor retardation precedes the onset of epileptic seizures or is presented without them. It is assumed that SETD1B dysfunction represents developmental encephalopathy with or without epilepsy.[1] SETD1B encodes histone H3 lysine 4 (H3K4) methyltransferase, which methylates histone H3 at the lysine-4 position as part of a multisubunit complex.[2] H3K4 state is associated with different locations in the genome and functional activity of genes.[2,3]
The SETD1B protein consists of 1966 amino acids and has several putative functional domains.[4] The N-terminus contains an RNA recognition motif (RRM 94-182 amino acid residues, 3 exons); in the middle part of the protein, there are two long anordered regions that differ in the conservative lysine–serine–aspartic acid motif and a helical structure.[2] At the C-terminus of SETD1B is the domain of the catalytic SET (1822–1948), which is crucial for the activity of histone methyltransferase. The N-SET domain (1668–1821) is located proximally, including the conservative motif (WIN).[5]
The literature we studied describes 49 cases with a mutation in the SETD1B gene and 3 cases with a deletion in chromosome 12 that includes this gene.[4,6-12]
In this report, we want to describe a new electroclinical phenotype associated with the previously unpublished variant c.4704dup (p.Ser161Gly) in exon 3 of the SETD1B gene, also called the RRM domain, containing an RRM.
CASE REPORT
Girl, 4 years old, from non-related healthy parents, up to the age of 4 months, developed according to age. At 4 months old, after recently suffering from an acute respiratory viral infection, she stopped holding her head and leaning on the arm. At the same time, there were short-term episodes of nodding movements of the head many times a day.
From the anamnesis vitae
The age of the mother at the time of the birth of the child was 38 years old.
At the age of 1 year and 6 months, short flinches and fading occurred. A night’s sleep was disrupted. Seizures were repeated many times up to 50 times a day. EEG with sleep monitoring revealed regional epileptiform activity in the frontal-central temporal regions with amplitude accent on the right, isolated in the right and left frontal temporal regions, and in the form of diffuse discharges. There was no obvious pathology on the brain MRI [Figure 1]. The first antiepileptic drug (AED) therapy with valproic acid was started at the age of 2. The frequency of seizures did not change. Then, topiramate was added to the treatment. Combined antiepileptic therapy had no positive effect. At the time of 2.5 years, the vocabulary consisted of 3–4 words, she could walk only with support, frequent falls, stumbling, and hypotension of the limbs. There are no changes in the dynamics of the EEG. Later, valproic acid was replaced with levetiracetam due to a hypersensitivity reaction. On the combination therapy, there was an improvement in the beginning of independent walking, a slight decrease in the frequency of myoclonic seizures, and the child became more active.
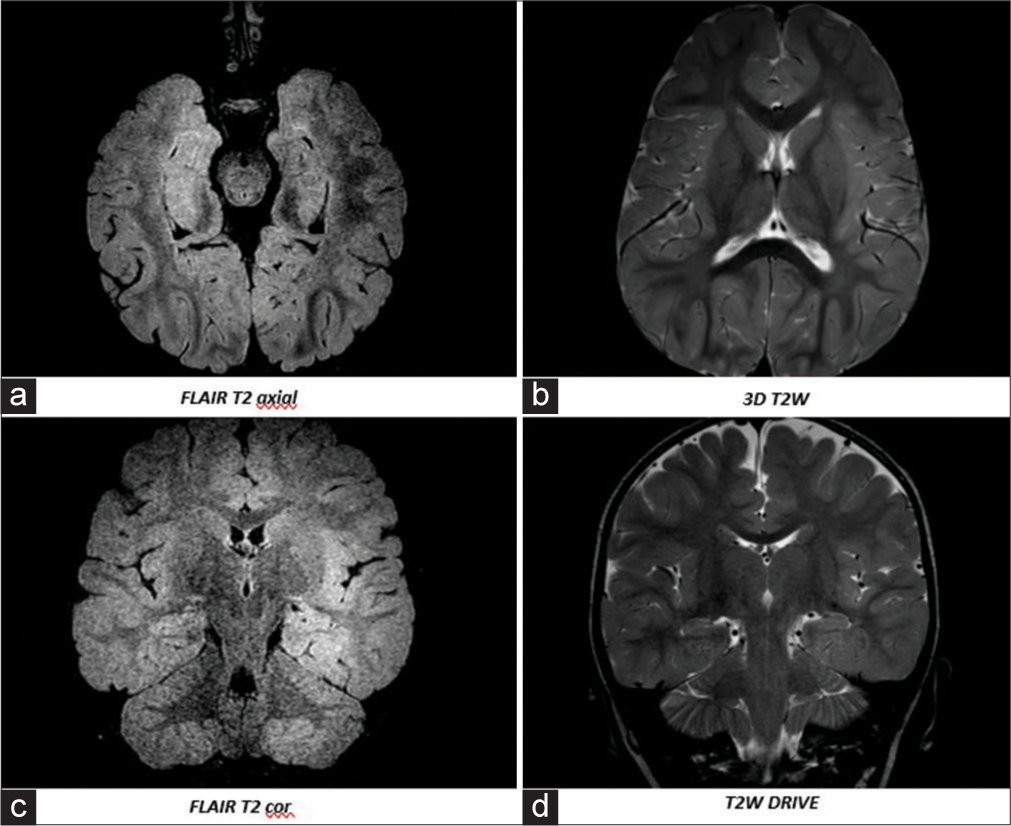
- Brain magnetic resonance imaging (HITACHI ECHELON 1.5 T from May 23, 2021 g (1 year and 8 months): (a, b) Axial images, (c, d) Coronal images. Gliotic changes in the frontal-parietal, periventricular, occipital regions, moderately pronounced external hydrocephalus.
A whole-exome sequencing detected a missense mutation in exon 3 of the SETD1B gene (NM_001353345.2:c.4704dupp.Ser161Gly), classified as a heterozygous probably pathogenic variant. This option was not described in the gnomAD and ClinVar population databases.
Further attempts to replace topiramate with zonisamide were made. However, she continued the previous AED (topiramate + levetiracetam) due to the development of an allergic reaction. We increased the dosage of levetiracetam.
During the last examination at the age of 4 years, seizures became less prolonged. Stabilization of the emotional background, formation of new skills, phrasal speech, and active socialization with other children were noted. There are complaints of episodes of fading with flinching up to 50 times a day, weakness after waking up, unsteady gait, frequent stumbling, falling, difficulty climbing stairs, obsessive movements of shaking her head, zipping, closing doors, hyperactivity, no sense of compassion other children, sleep disorder, rare episodes of autoaggression.
On examination
Weight is 19.7 kg (+1 SD). The skin is pale and seborrheic dermatitis is noted on the scalp. Flat feet, narrowed distal phalanges of the fingers, hypermobility of the joints. A high forehead, a prominent rounded tip of the nose, and thick eyebrows.
Neurological examination
She understands the spoken language and executes instructions partially. There is eye contact. There is no pointing gesture. Simple phrasal speech. Head circumference – 52 cm (+2 SD), macrocephaly. The gaze fixes and follows the subject periodically. Attention is quickly shifted to other subjects. Pharyngeal reflexes are revived. There is a decrease in muscle tone in the upper and lower extremities. Tendon reflexes are triggered, D = S. There is no paresis. She walks independently, her gait like myopathic. Govers’ symptom is negative. There is a urinary disorder.
According to the latest EEG [Figure 2], the epileptiform activity is the same.
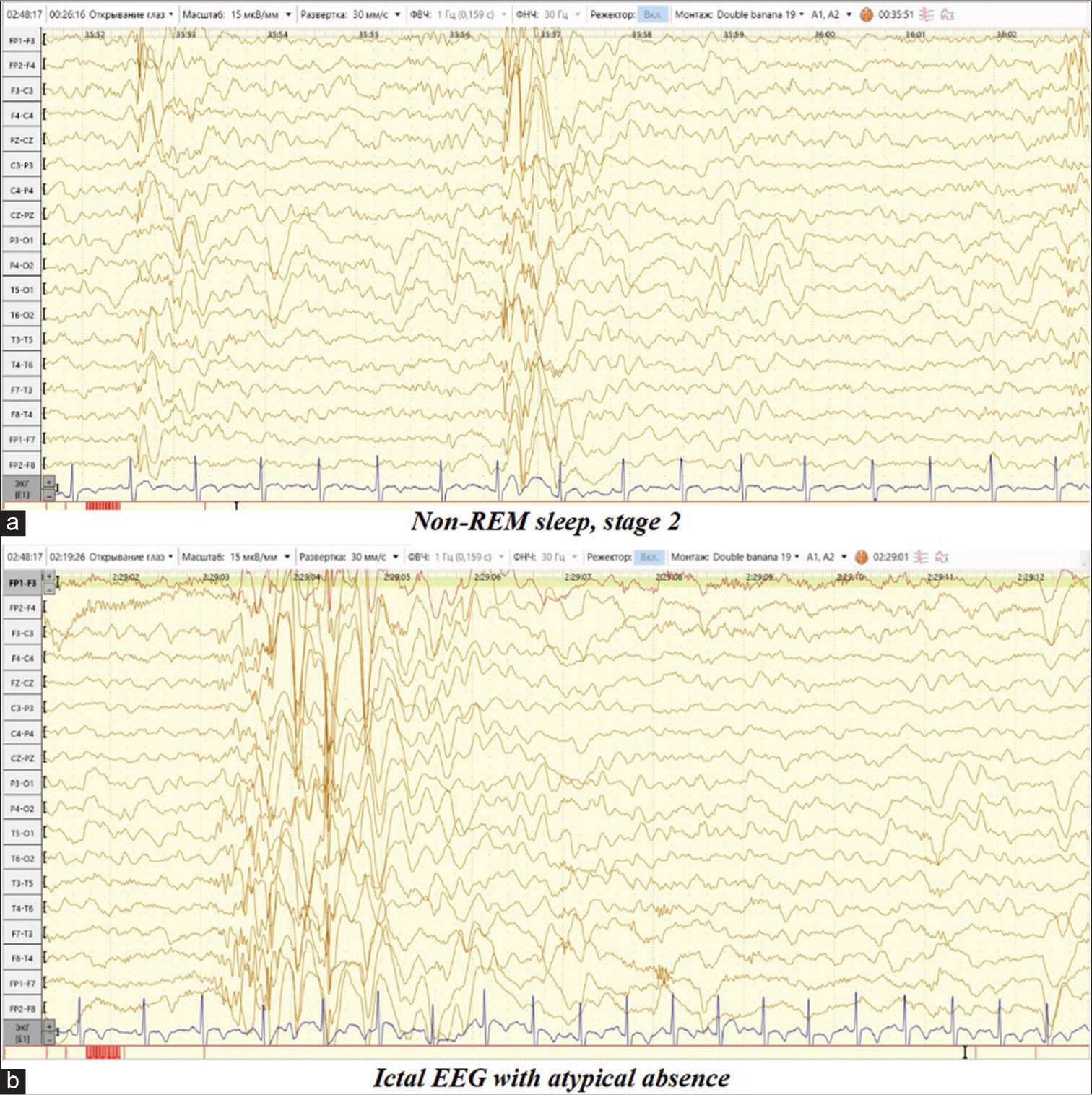
- Video electroencephalogram daytime sleep monitoring dated September 20, 2023. (a) Non-REM sleep, stage 2. Short diffuse discharges of complexes spike/double spike-slow wave with amplitude predominance over the frontal-central leads, passing into generalized discharges. (b) Ictal EEG: Complexes spike/double spike/polyspike-wave above the temporal leads bilaterally asynchronously with the capture of the frontal-central leads, followed by registration of a short bilateral synchronous discharge of high-amplitude complexes spike/double spike-slow wave with a frequency of 2–2.5 Hz., clinically accompanied by a short fading with a slow lowering of the head and body forward for up to 2 s.
DISCUSSION
We demonstrate a missense mutation in exon 3 of the SETD1B gene with such phenotypic manifestations as developmental delay, pharmacoresistant atypical absences and myoclonia, autism spectrum disorder, and hypotension. The phenotypic picture of our case is similar to 52 clinical cases with variants in the SETD1B gene previously described in the scientific literature. Most of the described mutations occur in the catalytic domains SET and N-SET. Moreover, there are only 6 cases in the RNA binding domain accounting for 3 exons. However, regardless of the location and type of mutation the phenotypic manifestations of the described cases are similar.
We also performed homologous modeling of the normal sequence (NM_001353345.2) with the implementation of pathological amino acid substitution obtained using artificial intelligence methods developed in the AlphaFold project from DeepMind. According to the results, a mutation in the RRM area leads to a stop codon and a shift in the reading frame, which in turn leads to the synthesis of a defect protein. However, the level of evidence of the result of our model is because we have no original primary data from the NGS laboratory for sequencing.
CONCLUSION
Our clinical report shows that the mutation in the SETD1B gene is clinically significant. Thus, the importance of genetic testing is emphasized, especially in cases with early onset of seizures in combination with developmental delay, pharmacoresistant epilepsy, and behavioral disorders. We want to pay the attention of the global scientific community for further research and a better understanding of the genotype-phenotypic correlation in SETD1B-associated nervous system disorders.
The authors confirm that all relevant consent forms were obtained from the patient’s parent for the publication of the clinical case. Consent was also given to the publication of clinical information in the journal. They were assured that the patient’s names and initials would not be published. Although every effort will be made to conceal the identity of the patient, complete anonymity cannot be guaranteed.
Ethical approval
The research/study was approved by the Institutional Review Board at Karaganda Medical University, No. 2, dated February 29, 2024.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship
Nil.
References
- SETD1B-related neurodevelopmental disorder In: Initial posting. Seattle (WA): University of Washington; 2022.
- [Google Scholar]
- Rbm15-Mkl1 interacts with the Setd1b histone H3-Lys4 methyltransferase via a SPOC domain that is required for cytokine-independent proliferation. PLoS One. 2012;7:e42965.
- [CrossRef] [PubMed] [Google Scholar]
- Active genes are trimethylated at K4 of histone H3. Nature. 2002;419:407-11.
- [CrossRef] [PubMed] [Google Scholar]
- Delineating the molecular and phenotypic spectrum of the SETD1B. Genet Med. 2021;23:2122-37.
- [CrossRef] [PubMed] [Google Scholar]
- Structural basis for WDR5 interaction (Win) motif recognition in human SET1 family histone methyltransferases. J Biol Chem. 2012;287:27275-89.
- [CrossRef] [PubMed] [Google Scholar]
- A genome-wide DNA methylation signature for SETD1B-related syndrome. Clin Epigenetics. 2019;11:156.
- [CrossRef] [PubMed] [Google Scholar]
- De novo variants in SETD1B cause intellectual disability, autism spectrum disorder, and epilepsy with myoclonic absences. Epilepsia Open. 2019;4:476-81.
- [CrossRef] [PubMed] [Google Scholar]
- A novel de novo frameshift variant in SETD1B causes epilepsy. J Hum Genet. 2019;64:821-7.
- [CrossRef] [PubMed] [Google Scholar]
- Epilepsy with eyelid myoclonia with atonic seizures and generalized paroxysmal fast activity: A novel electroclinical phenotype associated with SETD1B pathogenic variant. Epileptic Disord 2023:250-3.
- [CrossRef] [PubMed] [Google Scholar]
- Microdeletion of 12q24.31: Report of a girl with intellectual disability, stereotypies, seizures and facial dysmorphisms. Am J Med Genet A. 2015;167A:438-44.
- [CrossRef] [PubMed] [Google Scholar]
- An atypical 12q24.31 microdeletion implicates six genes including a histone demethylase KDM2B and a histone methyltransferase SETD1B in syndromic intellectual disability. Hum Genet. 2016;135:757-71.
- [CrossRef] [PubMed] [Google Scholar]
- Phenotypic spectrum of SETD1B-related disorder: Myoclonic absence seizures and concurrent intellectual disability-Insights from two cases. Seizure 2023:57-60.
- [CrossRef] [PubMed] [Google Scholar]




