
Translate this page into:
Clinical Spectrum of TGM6-Related Movement Disorders: A New Report with a Pooled Analysis of 48 Patients

Prateek Kumar Panda, DM Department of Pediatrics, Pediatric Neurology Division, All India Institute of Medical Sciences Rishikesh, Uttarakhand 249203 India drprateekpanda@gmail.com
This article was originally published by Thieme Medical and Scientific Publishers Pvt. Ltd. and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Abstract
Background Spinocerebellar ataxias (SCAs) are a diverse group of progressive neurodegenerative disorders. Until now, more than 20 genes have been implicated to be associated with this phenotype and TGM6 is one of these genes, associated with spinocerebellar ataxia-35 (SCA-35). The majority of disease-causing variants in the TGM6 gene predominantly have been reported from China and Taiwan and the association with Parkinson's disease (PD) have also been reported recently.
Methods We report the first Indian case with SCA-35 in a 16-year-old-boy with atypical age of onset at 9 years, prominent extrapyramidal features, intellectual disability, and a novel missense mutation in the TGM6 gene. We also reviewed and collated all previously published cases with pathogenic TGM6 variants.
Results Including the index case, 54 cases were identified from 10 relevant articles in literature and 48 cases had adequate clinical details to be included in the pooled analysis. Around two-thirds of reported cases had SCA-35 phenotype, with cerebellar atrophy. Onset in the majority of cases was the fourth decade of life onwards. A proportion of SCA-35 cases also had spasmodic torticollis, impaired proprioception, extrapyramidal features, and myoclonic jerks. The patients with PD had often early-onset milder symptoms, slower progression, and favorable response to levodopa/carbidopa. One patient each presented with episodic ataxia and dystonic tremor of the upper limb. Most of the cases had missense mutations, without any definite hotspot or genotype–phenotype correlation.
Conclusions TGM6 mutation should be suspected in patients with SCA like presentation, especially when it is accompanied by extrapyramidal features, spasmodic torticollis, impaired proprioception, or myoclonus.
Keywords
spinocerebellar ataxia
ataxia
TGM6
SCA-35
cerebellar ataxia

Introduction
Spinocerebellar ataxias (SCAs) comprise a diverse group of progressive neurodegenerative disorders and are considered as a subset of inherited cerebellar ataxias, with the autosomal dominant transmission. Core triad of symptoms in SCA patients includes gait ataxia and incoordination, dysarthria, and nystagmus/oculomotor apraxia.1 Affected patients in specific SCAs may manifest additional features like pyramidal or/and extrapyramidal signs, myoclonus, peripheral neuropathy, cognitive impairment, and ophthalmoplegia. CAG nucleotide repeat expansions are responsible for the majority of SCAs, which encode polyglutamine, and subsequently, produce toxic polyglutamine proteins (poly-Q).2 However, conventional mutations, such as point mutations, deletions, duplications, and frameshift mutations, are responsible for few other SCAs. SCA type 35 (SCA-35) is one such recently described SCA in the Han Chinese and Taiwanese population of East Asia, caused by mutations in the TGM6 gene.3 In recent years, TGM6 gene mutation has been detected in patients with Parkinson's disease (PD) of Chinese origin, episodic ataxia (EA), and dystonic tremors. Pathogenicity of reported mutations by functional assay, including their role in autophagy and α-synuclein production, has been demonstrated.4 5 6 7 8 9 10 11 12 However, outside the East Asia region, only a few cases of SCA-35 have been reported. We are describing the first case of Indian origin with TGM6 mutation of Indian origin with the SCA-35 phenotype. Unlike other reported cases, we had atypical onset from the first decade of life, significant extrapyramidal features, and a novel missense mutation. We also performed a pooled analysis of previously published individuals with TGM6 mutations in the existing literature. An institutional ethical clearance was taken for publishing this report.
Patient Characteristics
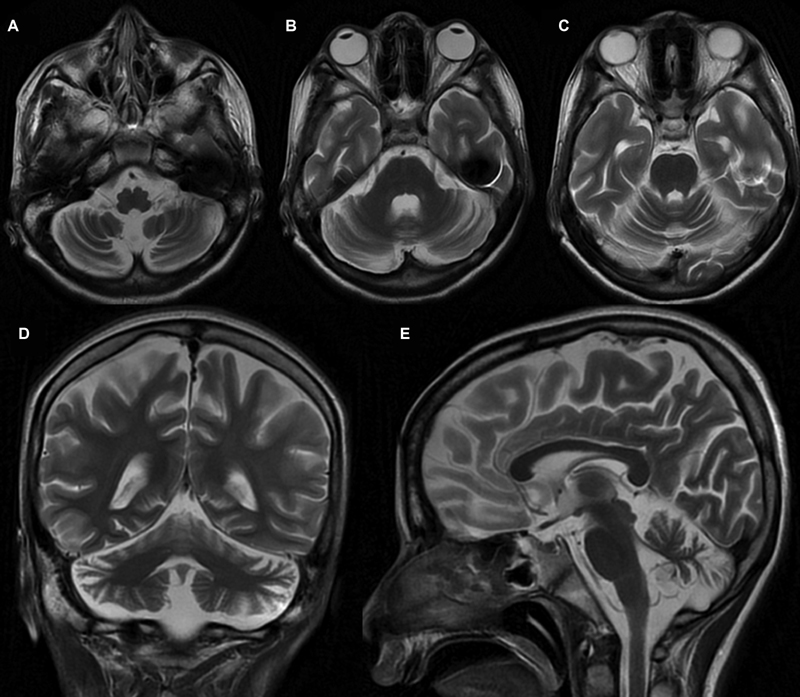
A 16-year-old boy firstborn to nonconsanguineous parents, without any adverse antenatal/perinatal events, presented with incoordination while walking and performing other tasks since 9 years of age. Initially, he had frequent falls with gait instability while walking. He had incoordination and often used to miss the target multiple times (undershooting and overshooting), even while approaching to the objects or doing any fine tasks like buttoning or unbuttoning the shirts or holding a pen. There was no history of any diurnal fluctuation of symptoms. For the previous 5 years, the parents observed that the child had tremulous movement of hands and fingers, increasing especially approaching the target object. Furthermore, he developed intermittent twisted posturing of hands for the previous 2 years and slurring of speech for the previous 4 years. On evaluation, the child was found to have appendicular and axial ataxia, dysmetria, dysdiadochokinesia, positive finger-nose test, heel-knee test, and brisk muscle stretch reflexes in lower limbs. He also had horizontal gaze nystagmus and oculomotor apraxia, with saccadic pursuits. Apart from intention tremor, he also had intermittent dystonic posturing of upper limbs. Neuroimaging of the brain revealed cerebellar atrophy (Fig. 1). Antibody against GAD65 and antitissue transglutaminase (TG) in the child was negative. His full-scale intelligence quotient (IQ) was 68, although his verbal IQ was better than his performance IQ, probably due to incoordination. His tandem mass spectrometry, urinary organic acid profile, serum prolactin, serum α-fetoprotein, immunoglobulin levels, blood, and cerebrospinal fluid lactate levels, and serum vitamin E levels were normal. Nerve conduction study and ophthalmological evaluation were also normal. He was started on trihexyphenidyl, levodopa/carbidopa, vitamin E, and different cofactors required for mitochondrial biosynthesis pathways without any observed clinical response. The informed consent of the parents was obtained for genetic analysis and publication of this report.
-
Fig. 1 Magnetic resonance imaging of the brain. T2-weighted axial (A–C), coronal (D), and sagittal (E) sequences showed bilateral cerebellar hemispheric and midline cerebellar atrophy.
Fig. 1 Magnetic resonance imaging of the brain. T2-weighted axial (A–C), coronal (D), and sagittal (E) sequences showed bilateral cerebellar hemispheric and midline cerebellar atrophy.
Genetic Testing
Genomic DNA was isolated from the patient sample and in situ hybridization of the coding exons and flanking intronic regions of the whole genomic DNA of the affected case was performed. Using next-generation paired read sequencing (using the Illumina NextSeq system, Eurofins-Emory Genetics Labs, United States), direct sequencing of the amplified and captured regions was performed. Low coverage regions, if any, are limited to 2 to 5% or less of the exons/nucleotides included in this panel. Sanger sequencing confirmation of the reported variants was also performed. Alignment to the reference human genome hg19/GRCh37 was performed using ECGIVarSure pipeline (ECGI [Eurofins Clinical Genetics India] bioinformatics pipeline) for data mapping, QC, variant calling, and annotation of variants with external and internal data sources. The analyzed region included the coding exons and ± 10bp of flanking intronic region on both sides of each exon, for each transcript listed. Certain deep intronic sites were also targeted. Only variants (SNVs [single nucleotide variants]/small indels) in the coding region and the flanking intronic regions (±10bp) with a minor allele frequency were reported.
Whole-exome sequencing detected a missense variant c.1876G > A (p.V626M) in the TGM6 gene (sequencing coverage of the coding region of the gene is 100%; located on chr20:2411582G > A; Read depth: 139x). The variant was absent in both parents and one available healthy male sibling, suggesting de novo origin and it was confirmed by Sanger sequencing about the presence of this variant in the affected case. Minor allele frequency of this mutation was found to be low in the general population (0.018% ExAC South Asian Frequency, 0.010% gnomAD South Asian Frequency, 0.00% ECGIdb). SCA-35 has been associated with mutations on this gene. Multiple lines of computational evidence (by PolyPhen-2, SIFT [sorting intolerant from tolerant], Mutation Taster2, Mutation Assessor, LRT [Likelihood ratio test]) supported a deleterious effect on the gene or gene product. This variant was classified as pathogenic according to the American College of Medical Genetics and Genomics guidelines for variant interpretation, and appropriate genetic counseling was offered to the parents.
Methods
Search Methods and Eligibility of Studies
In the databases of “PubMed, Scopus and Google Scholar,” a systematic literature review (only English language) was performed to find out the previously published individuals with pathogenic TGM6 variants using keywords for “TGM6” and “Spinocerebellar ataxia type-35” and “SCA-35” and “Parkinsonism” and “Cerebellar ataxia” that were in combination with study filters for original research, case series, and case reports. The primary objective of the review was to collate the pooled estimate of the clinical presentation of patients with SCA-35, harboring TGM6 variants. The review also intended to determine the clinical presentation of patients with TGM6 variants, presenting with parkinsonism and to determine abnormalities in neuroimaging and other diagnostic investigations, as well as the presence of any genotype–phenotype relationship in any of the two groups. Accordingly, the inclusion criteria we designed for the review were to include all articles describing at least one case with pathogenic TGM6 variant, irrespective of clinical presentation. The exclusion criteria of the review encompassed the articles describing only cases with variants of uncertain significance in TGM6 variants, inadequate clinical details, and TGM6 variants in nonhuman subjects. To find out the additional citations/manuscripts, we also reviewed the references of the published papers.
Selection of Studies and Data Extraction
All available full texts in literature, that is, papers, reports, and documents were taken. After screening the titles of documents, journal name, year of publication, and name(s) of the author(s), all duplicate items were removed. To select relevant articles and exclude nonrelevant ones, both authors (IKS and PKP) independently studied the articles carefully. Nonhuman studies and review articles were also excluded. On a predesigned case record form, clinical and molecular details of each case/s were noted and later on transferred to a Microsoft Excel spreadsheet.
Outcome Measures
The frequency of various clinical symptoms and signs in patients with SCA-35, apart from the distribution of various demographic variables, was taken as the primary outcome measure. Secondary outcomes were the distribution of those variables in patients with TGM6 mutations with parkinsonism. Other secondary outcome variables were the type and prevalence of various abnormalities in neuroimaging and other diagnostic tests, as well as the presence and type of genotype–phenotype correlations in these subjects.
In the patients with ataxia, Scale for the Assessment and Rating of Ataxia (SARA) was used to describe the severity of ataxia. First developed by Schmitz-Hübsch et al,13 this clinical scale evaluates impairments due to cerebellar ataxia in eight categories or items, with a minimum score being 0 (no ataxia) and a maximum score being 40 (most severe ataxia). The 8 items in this scale are gait, sitting, speech, stance, finger-chase test, heel-shin test, nose-finger test, and fast alternating movements. It is often used as an easily performed alternative to International Cooperative Ataxia Rating Scale, as its daily use is difficult many times due to a large number of assessment items.
Statistical Analyses and Data Synthesis
For categorical variables, frequencies and percentages were used. Standard descriptive statistics including means, standard deviation, median, and interquartile ranges were used for continuous variables. Student's t-test was used for the estimation of normal distribution variables and Wilcoxon rank-sum test was used for variables that did not follow the normal distribution for estimating the differences among comparison groups in continuous variables. Chi-squared test or the Fisher's exact test was used for assessing the differences in categorical variables. Using Statistical Package for the Social Sciences software version 26.0, all statistical analyses were performed.
Results
Twenty-three articles were selected from the systematic search. A total of 54 individuals (from 40 different families) with TGM6 variants were selected from 10 articles after excluding irrelevant articles and duplicate searches and including the clinical details of the case described above.3 4 5 6 7 8 9 10 11 12 Out of these, one was a whole-exome sequencing-based clinical study to identify novel variants responsible for SCA, with a paucity of adequate clinical details, and the six cases identified in this study with TGM6 missense mutations were variants of uncertain significance. Hence, the six cases described in this study were excluded from the final analysis. Out of the 48 patients included in the pooled analysis, 32 patients had SCA phenotype, one had EA2 phenotype, whereas the rest 15 patients of Chinese origin described in a recent study had parkinsonism, without any features of cerebellar ataxia.
Clinical Presentation
Cerebellar Ataxia Phenotype
In summary, out of 33 patients with cerebellar ataxia with TGM6 variants, 18 (54%) were male and the rest were female. Around half of the patients (48%) had onset in the fifth decade, while 12% of patients each had onset in the third, fourth, and sixth decades. Only two (6%) and three (9%) patients had onset of the illness in the first and second decades. The patients were diagnosed after a lag period of median 15 years of illness (range: 3–30 years). All patients had signs of persistent cerebellar ataxia, apart from the one with EA2 phenotype. He had episodes of ataxia lasting for hours together, with spontaneous resolution from 54 years of age and the episodes did not respond to acetazolamide. He also had nystagmus in between the episodes, but no residual truncal ataxia, migraine, or seizures. The demographic profile and clinical presentation of patients with spinocerebellar/EA have been described in Table 1.
|
Variable |
Frequency (n = 33) |
|---|---|
|
Males |
18 (54%) |
|
Females |
15 (46%) |
|
Age of onset |
|
|
First decade |
2 (6%) |
|
Second decade |
3(9%) |
|
Third decade |
4(12.1%) |
|
Fourth decade |
4(12.1%) |
|
Fifth decade |
16(48.4%) |
|
Sixth decade |
4(12.1%) |
|
Duration of illness in years (median, range) |
15 (3–30) |
|
Limb ataxia |
32(97%) |
|
Episodic ataxia |
1(3%) |
|
Dysarthria |
29(87%) |
|
Hand tremor |
27(81.8%) |
|
Nystagmus |
18(54%) |
|
Saccadic/pursuit aberrations |
8(24%) |
|
Spasmodic torticollis |
8(24%) |
|
Impaired proprioception |
6(18%) |
|
Myoclonus |
3(9%) |
|
Epilepsy |
1(3%) |
|
Extrapyramidal features |
4(12%) |
|
Parkinsonism |
2(6%) |
|
Dystonia |
4(12%) |
|
Brisk DTRs |
31(93%) |
|
SARA at examination |
17.56 ± 8.37 |
Abbreviations: DTRs, deep tendon reflexes; SARA, Scale for the Assessment and Rating of Ataxia.
Around 87, 81, and 54% of patients had dysarthria, hand tremor, and nystagmus. The average SARA score was 17.56 ± 8.37. Oculomotor apraxia was present in eight (24%) of cases, in the form of saccadic/pursuit aberrations. Other less frequent clinical features include spasmodic torticollis in eight (24%), impaired proprioception in six (18%), myoclonus in three (9%), epilepsy in one (3%), extrapyramidal features in three (9%), parkinsonism in two (6%), and dystonia in four (12%) patients. However, on clinical examination, 31 out of 33 patients had brisk deep tendon reflexes suggestive of pyramidal involvement. One patient presented at 55 years of age with isolated dystonic tremors of the right upper limb for 20 years, which subsequently involved voice and contralateral upper limb. He was tried with multiple medications without any sustainable response including the following drugs, that is, primidone, propranolol, trihexyphenidyl, gabapentin, levodopa, and topiramate. He did not show response to even deep brain stimulation of the ventral intermediate nucleus of the thalamus.
Parkinsonism Phenotype
Among the 15 patients, 9 (60%) were males, who presented with parkinsonism caused by TGM6 variants. A family history of PD was present in eight (53%) patients, although genetic testing was not available for the affected family members in any of the cases. The average age at onset of PD symptoms was 48.3 ± 13.9 years, whereas the average age at diagnosis was 52.7 ± 14.2 years, with an average disease duration of 5.5 ± 3.7 years. The clinical course of parkinsonism in these patients more or less resembled that of cases with idiopathic PD, but with milder symptom severity at onset and more insidious disease progression, over a longer duration. Clinical presentation and demographic profile of patients with parkinsonism phenotype have been described in Table 2.
|
Variables |
Frequency (n = 15) |
|---|---|
|
Male |
9(60%) |
|
Female |
5(33%) |
|
Age at diagnosis (y) |
52.7 ± 14.2 |
|
Age at onset (y) |
48.3 ± 13.9 |
|
Disease duration(y) |
5.5 ± 3.7 |
|
Family history |
8(53%) |
|
Initial presenting symptom |
|
|
Resting tremor |
8(53%) |
|
Bradykinesia |
7(47%) |
|
Prevalence of symptoms at latest follow-up |
|
|
Bradykinesia |
15(100%) |
|
Tremor |
14(93.3%) |
|
Rigidity |
12(80%) |
|
Postural instability |
5(33.3%) |
|
Olfactory disorder |
7(47%) |
|
REM sleep behavior disorder |
6(40%) |
|
Depression |
6(40%) |
|
Constipation |
9(60%) |
|
Sexual impotence |
2(13.3%) |
|
Sleep benefit in symptoms |
5(33.3%) |
|
Depression |
6(40%) |
|
Motor fluctuation |
3(20%) |
|
Beck depression inventory score |
9.8 ± 7.0 |
|
Sniffing Stick's test of olfactory function score (mean, SD) |
5.4 ± 2.3 |
|
Mini-Mental State Examination score (mean, SD) |
26.0 ± 5.2 |
|
Epworth sleepiness scale score (mean, SD) |
6.6 ± 5.3 |
|
Unified Parkinson disease rating scale score (mean, SD) |
23.3 ± 9.1 |
|
Stage of parkinsonism Hoehn and Yahr's scale (mean, SD) |
2.3 ± 0.7 |
Abbreviations: REM, rapid eye movement; SD, standard deviation.
Resting tremor and bradykinesia were the initial presenting symptoms in eight (53%) and seven (47%) patients, respectively. However, at the time of determination of genetic etiology, most of them had all classical features of parkinsonism, including bradykinesia in all patients, resting tremors in 14 (93.3%) patients, and rigidity in 12(80%) patients. Other less prevalent clinical features described in these patients were as follows: postural instability in five (33.3%), olfactory problems in seven (47%), rapid eye movement sleep-related behavior disorder in six (40%), depression in six (40%), constipation in nine (60%), sexual impotence in two (13.3%), sleep benefit in symptoms in five (33.3%), and motor fluctuation in three(20%) patients. The average Beck depression inventory score of PD patients was 9.8 ± 7.0, average Sniffing Stick's test of olfactory function score was 5.4 ± 2.3, Mini-Mental State Examination (MMSE) score was 26.0 ± 5.2, Epworth sleepiness scale score was 6.6 ± 5.3, unified PD rating scale score was 23.3 ± 9.1, and stage of parkinsonism according to Hoehn and Yahr's scale was 2.3 ± 0.7. Only one case had a significantly abnormal MMSE score. All patients had a favorable response to the levodopa/carbidopa combination. Pramipexole and trihexyphenidyl were also tried successfully in four and one patient, respectively.
Neuroimaging and Electrophysiological Study Findings
Magnetic resonance imaging (MRI) brain was performed in at least one proband from each of the kindreds with SCA-35. Cerebellar atrophy was present in all 17 cases, in whom MRI brain was performed, while two cases had additional atrophy of brain stem. MRI brain was normal in the patient who presented with EA phenotype. One case had subclinical sensory axonal neuropathy involving the sural nerve, along with abnormal somatosensory evoked potential. In contrast to this MRI brain had no significant abnormality in PD patients. But PET brain (dopamine transporter (DAT) imaging with [11C]-2β-carbomethoxy-3β-(4- fluorophenyl) tropane) in PD patients showed a marked reduction of DAT uptake in both caudate nuclei and putamen bilaterally in all probands and fluorodeoxyglucose-positron emission tomography (FDG-PET) brain showed increased metabolism in the thalamus, globus pallidus and putamen. Neuroimaging and mutational characteristics and other laboratory investigations of all 48 cases have been demonstrated in Table 3.
|
Investigation |
TGM6 variants with ataxia (n = 33) |
TGM6 variants with parkinsonism (n = 15) |
|---|---|---|
|
MRI brain |
Cerebellar atrophy in all 17 cases, in whom MRI brain was performed (except the one with episodic ataxia), 2 had also atrophy of brain stem |
Normal in all the family |
|
PET brain (DAT imaging with CFT) |
Not performed |
Marked reduction of DAT uptake in both putamen and caudate nuclei bilaterally in all probands |
|
FDG-PET brain |
Not performed |
Increased metabolism in the globus pallidus, thalamus, and putamen |
|
NCS |
Subclinical sensory axonal neuropathy (1) |
Not performed |
|
SSEP |
Abnormal (1) |
Not performed |
|
TGM6 mutation |
A missense mutation (29) Nucleotide deletion (1) Nucleotide duplication (2) Splice site mutation (1) |
A missense mutation (14) Splice site mutation (1) |
Abbreviations: CFT, [11C]-2β-carbomethoxy-3β-(4- fluorophenyl) tropane; DAT, dopamine transporter; FDG-PET, fluorodeoxyglucose-positron emission tomography; MRI, magnetic resonance imaging; NCS, nerve conduction study; SSEP, somatosensory evoked potential.
Genetic Mutations
Until now, 39 different variants in the TGM6 gene have been reported, out of which six are variants with uncertain significance and the rest were pathogenic variants. All of them were heterozygous, loss of function mutations with autosomal dominant transmission/de novo origin. One missense mutation c.331C > T (p.R111C) was found in one kindred each with parkinsonism and SCA-35 phenotype. One patient from each phenotype had a novel splice site mutation, while one and two patients with SCA-35 had nucleotide deletion and duplication, respectively. The rest of the 28 mutations, described in the remaining 43 patients, were also missense point mutations. However, for the site of these mutations, no hotspot on any of the exons of the TGM6 gene could be identified. No definite genotype–phenotype correlation could be established from the details of available patients. No statistically significant difference could be elicited similarly between the age of onset (p = 0.43), age of presentation (p = 0.36), gender distribution (p = 0.93), or mutation characteristics (p = 0.56) between the patients presenting with SCA-35 or PD. Furthermore, the clinical characteristics of males and females with either phenotype were not significantly different in any prospects.
Discussion
We have described the detailed clinical features of 48 cases with TGM6-related movement disorders. Around two-third of cases presented with cerebellar ataxia, whereas the rest of the patients presented with features of parkinsonism. While most patients had onset in the young adult age group with SCA-35 phenotype, these two contrasting phenotypes do not appear to be completely watertight. Few patients with SCA-35 had variable extrapyramidal features, including parkinsonism. However, FDG or DAT PET scan was not performed in any of these patients, which could have identified subclinical abnormalities. If such subclinical abnormalities are detected in the future in the majority of cases of SCA-35, then probably levodopa/carbidopa combination should be tried in them as one of the potential therapeutic options. Why some of the missense point mutations lead to SCA phenotype with cerebellar atrophy, whereas, rest of the mutations lead to late-onset PD phenotype, remains unanswered and needs to be investigated in future studies. Recently, few affected cases in kindred with CAG repeats in the ATM gene and additional variants in the TGM6 gene. Although the interaction between these two genes needs to be examined carefully in the future, another task for clinicians will be to perform linkage analysis and explore other trinucleotide repeats in patients with SCA-35 phenotype, having TGM6 variants. Fung et al14 have also recently cast doubt on the pathogenicity of TGM6 variants in SCA-35 patients. Since SCA-35 is only a recently described entity, molecular testing based on large population screening studies in patients with unexplained ataxia will only generate concrete evidence about the true pathogenic role of TGM6 in SCA patients.
Characteristics of TGM6 Gene and the Encoded Protein
TGM6 gene (OMIM#613900) located on chromosome 20 encodes for a protein named TG6, a member of the TG family. TGs are Ca2+ dependent enzymes, which catalyze transamidation reactions to form intra or intermolecular isopeptide bonds between glutamine and lysine residue and help in covalent cross-linking of the protein.15 TGM6 is expressed in basal ganglia and cerebellum, especially in Purkinje cells, thereby explaining ataxia and parkinsonism in patients with pathogenic TGM6 variants.3 4 12
TG proteins comprise four contiguous ellipsoidal domains: one catalytic core domain with enzymatic activity, one aminoterminal β-sandwich domain, and two carboxy-terminal β-barrel domains that regulate the enzymatic activity.16 As reported pathogenic mutations have been documented in all four domains; it seems there is no hotspot for mutations, and alteration in any of the domains can produce deleterious effects on TG6 activity.17
Underlying Pathogenetic Mechanisms
Polyglutamine Aggregation Deposition
Apart from TG6, some of the other TG proteins have also been suggested in the pathogenesis of various other neurodegenerative diseases. TG6 and TG2 proteins convert soluble forms of poly-Q into an insoluble form, thereby promoting poly-Q aggregate formation. Apart from SCAs that are well known to be poly-Q diseases, other neurodegenerative diseases like Alzheimer's disease, Huntington disease, and idiopathic parkinsonism have also been postulated to be caused by aberrant or reduced expression of TGs.3 7 12
Disruption of Autophagy and α-Synuclein Deposition
TGM6 variants cause parkinsonism by disrupting the normal autophagy process and reducing metabolism of α-synuclein. TG2 has been shown to catalyze α-synuclein crosslink formation, which is one of the key initial steps in the pathogenesis of PD. The in vitro studies on A53T-SH-SY5Y cells prepared as PD models showed consistent results. While the cells overexpressing wild-type (WT) TG protected the cells by enhancing autophagy and reducing α-synuclein, this protective effect was found to be attenuated in cells with mutant TG6. Functional TG activity grossly reduces in cells with mutated TGM6. These recently revealed novel pathogenetic mechanisms delineate the correlation between TGM6 and parkinsonism and provide a potential therapeutic target for PD.4
Resemblances with Gluten Ataxia
In patients with celiac disease, anti-TG6 antibodies are associated with gluten ataxia; and ataxic symptoms could be reproduced, when anti-TG2/3/6 cross-reactive antibodies or single-chain variable fragments are injected intraventricularly in mice. None of the patients with TGM6 variants have been demonstrated to have anti-TG6 antibodies. In celiac disease, gluten-specific CD4 T cells drive autoantibody production, hence removing gluten is an effective treatment through preventing T cell help to B cells. However, in genetic TG6 deficiency this most probably has no role to play.6 Larger studies with longer follow-up periods are required to demonstrate whether any true resemblance exists between these two illnesses.
Unfolded Protein Response
Tripathy et al9 in 2017 analyzed in vitro pathogenicity of 12 reported TGM6 variants, proposed to be linked with SCA-35, including seven novels and five previously reported mutations, and found another novel mechanism to explain the molecular basis behind neurotoxicity caused by mutant TG6. Predominantly in the nucleus and perinuclear area, the WT-TG6 could be localized. In cells with mutated TG6, there was loss of enzymatic function and insolubility of TG6 mutants, leading to nuclear depletion of TG6 and aberrant accumulation in the perinuclear area. An unfolded protein response gets activated by the increased and abnormal accumulation of mutated TG6 proteins in the perinuclear area. This endoplasmic reticulum stress response activated by mutated, nonfunctional TG6 proteins with abnormal conformation is the leading cause behind neuronal death and this has been shown in animal models of SCA-35 in drosophila melanogaster.9
Sensitization of Neurons to Apoptosis
Guan et al18 have demonstrated that overexpression of the TG6 mutants increases the activity of caspases and sensitizes cells to staurosporine-induced apoptosis, apart from exhibiting reduced TG activity and stability. The pathogenesis of SCA-35 might be related to the proapoptotic role of TG6 mutants.
Pathogenesis behind Episodic Ataxia Phenotype
But how TG6 mutants led to EA phenotype in one patient remains unclear. Progressive cerebellar ataxia is classically seen in SCAs; however, some subtypes like SCA-6 and SCA-27 may present with fluctuating ataxia and resemble EA2. Both SCA-6 and EA2 are allelic disorders, caused by mutation in CACNA1A gene. Furthermore, within the same family, different affected members can have diverse phenotypes ranging from SCA-6 to EA2.19 Mutations in FGF14, responsible for SCA-27, although classically causes autosomal dominant EA, may rarely cause fever triggered EA.20
Potential Therapeutic Targets
The TG pathway, poly-Q aggregate formation, disruption of autophagy, α-synuclein deposition, unfolded protein response, the resemblance with gluten ataxia, fluctuating ataxia, and dystonia in some affected cases and favorable response to levodopa/carbidopa and pramipexole in TGM6 mutants with PD are all promising potential therapeutic targets.4
Clinical Presentation
The pooled analysis showed TGM6 variants caused predominantly two diverse phenotypes, one presenting as SCA and another with PD, often early-onset and slowly progressive, as compared with the other patients with PD. Unlike our case, most patients with TGM6 variants were found to have onset after the fourth decade, in both SCA and parkinsonism subgroup.3 4
Characteristics of Patients with SCA-35 Phenotype
In patients with SCA, presenting symptoms included insidiously progressive gait ataxia, pursuit and saccadic aberrations, dysarthria, action tremor, and pyramidal/extrapyramidal signs in a few cases. The exact description for the neuroimaging discrepancy between both phenotypes remains inconclusive. However, most patients with idiopathic parkinsonism also demonstrate grossly normal neuroimaging in the early stages, whereas a significant proportion of patients with other types of SCA also have cerebellar atrophy.3 4 5 6 7 8 9 10 11 12
SCA-35 was first reported by Wang et al3 in 2010 in a four-generation Han Chinese family, caused by mutations in the TGM6 gene, encoding for TG6. They utilized both whole-exome sequencing and linkage analyses together in four patients to identify this novel gene probably responsible for ataxia in human beings. They identified a missense variation, c.1550T-G transition (L517W), involving exon 10 of the TGM6 gene. They proposed that this mutation had a significant functional impact in bioinformatics study, although it was later contradicted by Thomas et al17). This mutation was, however, at a highly conserved position and completely cosegregated with the SCA phenotype in the fourth-generation family. The mutation was also located in 20p13–12.2, the same region identified by linkage analysis. They found further evidence that TGM6 mutations can cause SCA in human beings when they found another pathogenic missense variant cosegregating with the phenotype in an additional Chinese family with SCA.21 This second single-nucleotide substitution mutation, D327G, was found to interfere with Ca2+ binding at site 3 in structural remodeling and therefore may have compromised enzyme activation. But the mechanism by which the first mutation caused the deleterious effect remained obscure. Haploinsufficiency has been proposed as one possible mechanism by Thomas et al,17 by which mutation of a single allele can produce the dominant ataxia phenotype, but they also proposed that difficulties in the folding of mutant proteins leading to neurotoxicity could be another possible mechanism.
Subsequently, two additional Chinese families with SCA-35 were reported, harboring TGM6 pathogenic variants. However, the search for TGM6 mutations in 118 Caucasian cases of sporadic SCA did not yield any positive results. But similar search in 109 molecularly unassigned probands from Taiwanese SCA families, in whom underlying etiology remained unexplained, yielded three distinct TGM6 mutations, out of which two were novel mutations, like our case. Another study estimated that SCA-35 constituted 0.6% of patients with SCA phenotype from the Han-Chinese population of Taiwanese descent.5 7
Initially, SCA-35 was considered to be a disease of elderly adults universally, with onset from the fifth decade of life onwards. Later, cases with much younger-onset and even two adolescents affected with SCA-35 were reported. The case we presented is atypical not only because of the first reported case from India with a novel missense mutation but also due to the younger age of onset in the first decade of life. Despite all these previous cases, it is notable to mention that the role of TGM6 mutations accompanying potential early-onset SCA-35 is not well supported in a recent article, as the presence of this mutation poorly correlates with what is seen in cases where linkage has been more formally established and also in segregation analysis in affected family members in a report by Chen et al.22 Moreover, the mutation reported in this case report was the same L517W variant, reported initially by Wang et al.3 Although the affected family members in this report had CAG expansions in the ATN1 gene, the possible interaction between ATN1 and TGM6 cannot be ruled out completely.
As we have previously described, early manifestations are nonspecific and the same as for other SCAs including gait difficulties, dysarthria, and hand tremors. But spasmodic torticollis, impaired proprioception, and slow ocular pursuits are some of the relatively specific features, which may help clinicians in suspecting SCA-35 in patients with SCA phenotype. However, these findings have been reported only in a minority of affected patients. Another peculiar feature found in SCA-35 patients with onset in the first decade or adolescence is cognitive impairment (as found in our case), also seen in some other SCAs (SCA-2, -8, -13, -17, -19/22, -21, -36, and -44).1 Future studies are likely to report more distinct features and broaden the clinical spectrum of this illness.
Characteristics of Patients with Parkinson's Disease Phenotype
Compared with PD patients without any definite monogenetic abnormality, early onset PD patients with TGM6 variants exhibited milder symptoms for a longer duration, with slower disease progression. PET imaging findings in patients with PD caused by TGM6 variants resembled other PD patients. Although these two diverse, apparently mutually exclusive phenotypes caused by TGM6 variants were attributed to genetic heterogeneity, this might not be the true cause, as there are four cases of SCA-35, as described above, who also demonstrated some extrapyramidal features, like dystonia and parkinsonism. However, the reverse clinical picture has not been reported until now, as all described cases of PD did not have any evidence of significant cerebellar involvement.4
At the same time, it is noteworthy to mention that few families having molecularly confirmed patients with other SCA mutations (SCA-2 and -3) demonstrated pure parkinsonism or parkinsonism predominant phenotype. SCA-2 family with six affected individuals showing pure parkinsonism phenotype was described by Sun et al21 along with two other individuals with cerebellar ataxia.
Furtado et al23 have described another SCA-2 family in 2004 with PET brain imaging showing abnormalities similar to results found in idiopathic PD patients. Bettencourt et al24 and Buhmann et al25 have described SCA-3 patients initially presenting with parkinsonism and responding favorably to levodopa. However, they manifested cerebellar signs too after several years of the disease progression. Further prolong monitoring in the reported parkinsonism patients with TGM6 variants might show the development of cerebellar signs.
Population Studies and Prevalence of TGM6 Variants
The TGM6 variants that have been demonstrated in functional assays to be pathogenic were included in our pooled analysis and those belonged to China, India, Europe, Puerto Rico, etc., showing the cross-cultural existence of pathogenic TGM6 mutations. As most of the TGM6 mutations documented in SCA-35 and PD patients are pathogenic in situ, not shown in healthy, ethnically matched controls, and cellular studies showed adverse biological effects, thus the pathogenic role of TGM6 variants in humans is highly likely. Most of the mutations reported in the TGM6 gene are missense mutations, although rarely reported are the intragenic deletion, duplication, and even splice site mutation. However, a genotype–phenotype relationship could not be established, as the patients with few variants have been found to present with both SCA and early-onset PD phenotype, as described above.3 4 5
Despite all these, Fung et al14 cast doubt upon TGM6 mutations causing SCA-35 phenotype, as they identified eight additional Chinese families with TGM6 variants, without any clinical features of SCA-35. They opined that many of the missense or loss of function variants in the TGM6 gene are likely to be permitted. Hence, careful interpretation of pathogenicity analysis by bioinformatics and genotype–phenotype matching is highly essential.26 27
Our review has several limitations. Most studies are small case series only, which used various description and quantitative measurement terminologies, which often resulted in difficulty while collating for determining pooled estimate. All of the cases have also not performed proper linkage analysis and functional assays to truly describe the pathogenicity of detected variants. In some population-based studies, often we were unable to detect the detailed information regarding various clinical parameters. Hence, in all probability, for various parameters, the described values in the result section may only be a crude approximation rather than the exact value. Six cases of TGM6 variants that were excluded as variant of uncertain significance might be pathogenic in reality, as functional assays were not performed in them. Nerve conduction study, detailed neuropsychiatric assessment, and even neuroimaging findings were not available in all patients with SCA-35 phenotype. Significant publication bias could also have compromised our study results, as cases with a positive result in genetic reporting are more likely to be published. Despite the above limitations, this is the first attempt to date that provides an overview of the clinical spectrum of patients with TGM6 variants. In the future, this review is likely to help clinicians in planning systematic evaluation of patients with suspected/confirmed SCA-35 phenotype or PD patients with TGM6 variants. It will also make aware the clinicians to test for TGM6 mutations in patients with unexplained ataxia and idiopathic PD phenotype.
Conclusion
In patients with SCA like presentation with the probable autosomal dominant transmission, especially when it is accompanied by extrapyramidal features, spasmodic torticollis, impaired proprioception, or myoclonus, TGM6 mutation should be clinically suspected. Although most patients had onset after the third decade of life, presentation in childhood or adolescence should not preclude clinicians from suspecting SCA-35.
Conflict of Interest
None declared.
Funding None.
References
- TGM6 identified as a novel causative gene of spinocerebellar ataxias using exome sequencing. Brain. 2010;133:3510-3518. (Pt 12):
- [Google Scholar]
- TGM6 variants in Parkinson's disease: clinical findings and functional evidence. J Integr Neurosci. 2020;19(1):51-64.
- [Google Scholar]
- Spinocerebellar ataxia 35: novel mutations in TGM6 with clinical and genetic characterization. Neurology. 2014;83(17):1554-1561.
- [Google Scholar]
- Hispanic spinocerebellar ataxia type 35 (SCA35) with a novel frameshift mutation. Cerebellum. 2019;18(2):291-294.
- [Google Scholar]
- Whole exome sequencing identifies a novel mutation in the transglutaminase 6 gene for spinocerebellar ataxia in a Chinese family. Clin Genet. 2013;83(3):269-273.
- [Google Scholar]
- Genetic variants associated with episodic ataxia in Korea. Sci Rep. 2017;7(1):13855.
- [Google Scholar]
- Mutations in TGM6 induce the unfolded protein response in SCA35. Hum Mol Genet. 2017;26(19):3749-3762.
- [Google Scholar]
- SCA 35 presenting as isolated treatment-resistant dystonic hand tremor. Parkinsonism Relat Disord. 2017;37:118-119.
- [Google Scholar]
- Exome sequencing in the clinical diagnosis of sporadic or familial cerebellar ataxia. JAMA Neurol. 2014;71(10):1237-1246.
- [Google Scholar]
- TGM6 gene mutations in undiagnosed cerebellar ataxia patients. Parkinsonism Relat Disord. 2018;46:84-86.
- [Google Scholar]
- Scale for the assessment and rating of ataxia: development of a new clinical scale. Neurology. 2006;66(11):1717-1720.
- [Google Scholar]
- A significant inflation in TGM6 genetic risk casts doubt in its causation in spinocerebellar ataxia type 35. Parkinsonism Relat Disord. 2019;63:42-45.
- [Google Scholar]
- Transglutaminase-6 is an autoantigen in progressive multiple sclerosis and is upregulated in reactive astrocytes. Mult Scler. 2017;23(13):1707-1715.
- [Google Scholar]
- Evolution of transglutaminase genes: identification of a transglutaminase gene cluster on human chromosome 15q15. Structure of the gene encoding transglutaminase X and a novel gene family member, transglutaminase Z. J Biol Chem. 2001;276(35):33066-33078.
- [Google Scholar]
- Transglutaminase 6: a protein associated with central nervous system development and motor function. Amino Acids. 2013;44(1):161-177.
- [Google Scholar]
- Spinocerebellar ataxia type 35 (SCA35)-associated transglutaminase 6 mutants sensitize cells to apoptosis. Biochem Biophys Res Commun. 2013;430(2):780-786.
- [Google Scholar]
- Episodic ataxia and SCA6 within the same family due to the D302N CACNA1A gene mutation. J Neurol Sci. 2016;371:81-84.
- [Google Scholar]
- A novel frameshift mutation in FGF14 causes an autosomal dominant episodic ataxia. Neurogenetics. 2015;16(3):233-236.
- [Google Scholar]
- Genetic and clinical analysis in a Chinese parkinsonism-predominant spinocerebellar ataxia type 2 family. J Hum Genet. 2011;56(4):330-334.
- [Google Scholar]
- TGM6 L517W is not a pathogenic variant for spinocerebellar ataxia type 35. Neurol Genet. 2020;6(3):e424.
- [Google Scholar]
- Profile of families with parkinsonism-predominant spinocerebellar ataxia type 2 (SCA2) Mov Disord. 2004;19(6):622-629.
- [Google Scholar]
- Parkinsonian phenotype in Machado-Joseph disease (MJD/SCA3): a two-case report. BMC Neurol. 2011;11:131.
- [Google Scholar]
- Dopaminergic response in Parkinsonian phenotype of Machado-Joseph disease. Mov Disord. 2003;18(2):219-221.
- [Google Scholar]
- COL6A3 mutation associated early-onset isolated dystonia (DYT)-27: report of a new case and review of published literature. Brain Dev. 2020;42(4):329-335.
- [Google Scholar]
- Response to carbamazepine in KCNQ2 related early infantile epileptic encephalopathy. Indian J Pediatr. 2019;86(3):301-302.
- [Google Scholar]




