
Translate this page into:
Clinical Course, Imaging Characteristics, and Therapeutic Response in Myelin Oligodendrocyte Glycoprotein Antibody Disease: A Case Series
This article was originally published by Thieme Medical and Scientific Publishers Private Ltd. and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Abstract
Myelin oligodendrocyte glycoprotein (MOG) antibody disease is a novel central nervous system autoimmune disorder which forms part of aquaporin 4 (AQP-4) negative, neuromyelitis optica (NMO) spectrum disorder. It has a distinct clinical profile, neuroimaging features and courses from AQP-4 positive NMO and multiple sclerosis. This article is a case series of six patients with MOG antibody disease with longitudinal follow-up for up to 8 months.
Keywords
India
magnetic resonance imaging
myelin oligodendrocyte glycoprotein
neuromyelitis optica
rituximab

Introduction
Myelin oligodendrocyte glycoprotein (MOG) antibody disease is a novel central nervous system (CNS) autoimmune disorder. MOG is a component of outermost sheath of myelin and oligodendrocyte membrane.1 It serves functions like oligodendrocyte microtubule stability, cell adhesion molecule, and myelin-immune system interactions.2 Even though, MOG is a minor component of myelin sheath (0.05%), it is highly immunogenic. IgG1 subclass of antibodies directed against MOG, has been found in experimental autoimmune encephalomyelitis.3 In clinical practice, MOG antibodies have been found in 36.4% cases of acute disseminated encephalomyelitis (ADEM), 26.9% cases of aquaporin-4 negative neuromyelitis optica spectrum disorder (NMO-SD), and 5.1% cases of multiple sclerosis.4 MOG antibody disease in adults is thought to be a part of NMO-SD, with a milder phenotype than seropositive neuromyelitis optica (NMO) and distinct clinical and radiological features.5 There are limited data about the clinical profile of MOG antibody disease from India. This article describes the clinical profile, neuroimaging characteristics, treatment response, and short-term outcome in six adult patients with MOG antibody disease from a tertiary care center in South India.
Methods
This was a single-center, unblinded, prospective study. All consecutive patients with clinically suspected CNS demyelination, from the department of neurology, were included. Serum samples of these patients were tested for IgG-MOG antibody and IgG anti-aquaporin-4 antibody using a cell-based immunoassay using transfected cell lines in a semiquantitative method.6 All patients underwent neurological examination by at least two consultants, independently. Relapses were calculated with annualized relapse rate and disability was assessed using expanded disability status scale (EDSS). Cerebrospinal fluid (CSF) analysis, visual evoked potentials (VEP), contrast magnetic resonance imaging (MRI) of brain and spinal cord, erythrocyte sedimentation rate (ESR), and antinuclear antibodies (ANA) were planned in all patients. All patients were treated with pulse methylprednisolone (1,000 mg intravenous for 3–5 days followed by oral steroids at 1 mg/kg in a standard tapering regimen), followed by rituximab (induction with 375 mg/m2 every week for four doses, followed by maintenance dose of 375 mg/m2 every 6 months). Patients were longitudinally followed up for remission, disease progression, or relapse.
Results
During the period from February 2018 to September 2018, 47 patients with clinically suspected CNS demyelination were tested for IgG MOG-antibody. Six patients (12.8%) were tested positive.
Case 1
A 30-year-old female presented with lower abdominal pain followed by acute urinary retention, without any limb weakness or sensory symptoms. She had a history of short febrile illness 2-weeks back. Neurological examination revealed Grade 5/5 power of all limbs, with bilaterally exaggerated deep tendon reflexes in upper and lower limbs, extensor plantar responses and absent abdominal reflexes, without exaggerated jaw jerk with intact sensations and a normal gait. The EDSS score was 4 (on a scale 0–15, higher values indicating more severe disability). CSF study showed 71 cells (97% lymphocytes), positive oligoclonal band (OCB), (serum OCB was also positive). Urodynamic studies showed detrusor-sphincter dyssynergia. MRI of the spine showed multiple ill-defined patchy lesions extending from cervical to conus segments. She received pulse methylprednisolone followed by rituximab induction. She received her first maintenance dose of rituximab 6 months later. On her last follow-up after 34 weeks, she had no urinary symptoms, and EDSS had improved to 2.
Case 2
A 35-year-old male presented with acute onset pain in the left eye followed by blurring of vision. He had history of bilateral optic neuritis in 2002 (which recovered fully with steroids) and 2009 (with good recovery in the left eye, but poor vision in the right eye). On examination, he had a visual acuity 0.05 in the right eye and 0.3 in the left eye, with sluggish direct light reflexes and a relative afferent pupillary defect (RAPD), in the right eye, and optic disc pallor in the right eye. His deep tendon reflexes were exaggerated, without sensory symptoms or signs and a normal gait. EDSS at onset was 4. VEP showed no response from either eye. He received pulse steroids, followed by rituximab. On the last follow-up at 28 weeks, he had no further relapses, EDSS improved to 3, visual acuity in the left eye improved to 0.8, but the right eye remained unimproved.
Case 3
A 39-year-old female presented with pain in the left eye followed by blurring of vision. She had a history of left optic neuritis 6-months back, which recovered after pulse steroids. On examination, the visual acuity was 0.7 in the left eye and normal in the right, with RAPD and pale optic disc in the left eye. Rest of the neurological examination was normal. EDSS at the onset was 1. VEP showed prolonged P100 latency in the left eye and was normal in the right. Her ESR was 20 mm/1st hour, ANA and anti-Sjögren’s syndrome type-B (SSB) were positive. CSF study showed protein of 86 mg/dL, without pleocytosis. MRI brain revealed two dot-like fluid-attenuated inversion recovery (FLAIR) hyperintense lesion in the subcortical white matter of right frontal lobe. After pulse steroids, she received rituximab induction. At 26-week follow-up she had no further relapses, visual acuity in the left eye had improved to 0.9 and EDSS was 1.
Case 4
A 25-year-old male presented with a relapsing remitting illness characterized by recurrent blurring of vision in the right eye. The first attack was in September 2017. Subsequently he had three relapses in the same eye in March, April, and July 2018. On examination, visual acuity was 0.3 in the right eye and 0.8 in the left, with RAPD and a pale optic disc in the right eye. Rest of the neurological examination was unremarkable. EDSS at the onset was 3. VEP showed absent response in the right eye and prolonged P100 latency in the left eye. CSF study was unremarkable. MRI brain revealed a faint enhancing hyperintensity near the right side of optic chiasma. He received pulse steroids with rituximab. At 11-week follow-up, he had no further relapses, his visual acuity had improved to 0.8 in both eyes and EDSS was 1.
Case 5
A 57-year-old male presented with a chronic progressive visual loss in the right eye of 4-years duration, without diplopia, sensorimotor symptoms in the limbs, or similar complaints in the left eye. On examination, visual acuity was 0.1 in the right eye and 0.3 in the left eye, with RAPD and primary optic atrophy of the right eye. Rest of the cranial nerves were normal. He had grade 5/5 power of all limbs, with exaggerated deep tendon reflexes bilaterally, with bilateral flexor plantar response. EDSS was 3. VEP showed prolonged P100 latencies in both eyes. MRI showed FLAIR hyperintense lesion in the periventricular white matter of left occipital lobe (Fig. 1). He was treated with pulse steroids, followed by rituximab. On follow-up after 20-weeks, he had no further relapses or progression of disability (EDSS remained 3) and visual acuity improved to 0.5 in the left eye, however, the acuity in right eye did not improve.
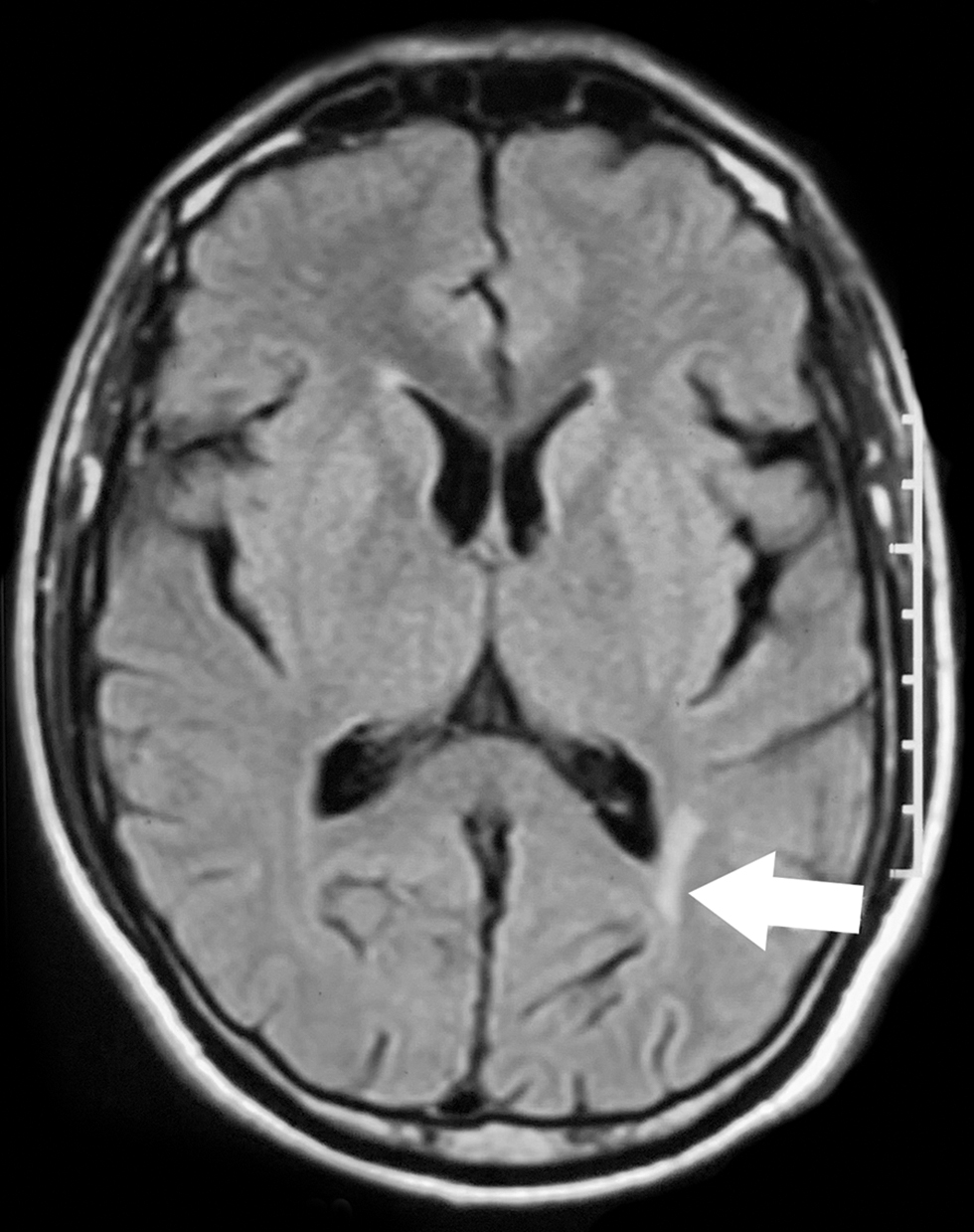
-
Fig. 1 Axial T2 FLAIR MRI showing hyperintense lesion in periventricular white matter of left occipital lobe (arrow). FLAIR, fluid-attenuated inversion recovery; MRI, magnetic resonance imaging.
Fig. 1 Axial T2 FLAIR MRI showing hyperintense lesion in periventricular white matter of left occipital lobe (arrow). FLAIR, fluid-attenuated inversion recovery; MRI, magnetic resonance imaging.
Case 6
A 36-year-old female presented with short duration neurological illness which started as sensory symptoms over right half of the face and body, followed by weakness of both lower limbs and sensory symptoms below the trunk, few hours later. She developed acute urinary retention and constipation the next day. She did not have any visual complaints. On examination, she had a jerky nystagmus on looking to the left, with hemisensory impairment over face. She had grade 4/5 power in both lower limbs, with normal power of upper limbs, exaggerated deep tendon reflexes bilaterally in upper and lower limbs with bilateral extensor plantar, and impaired touch and vibration sense below D6 level. EDSS was 6. CSF study and VEP were normal. MRI showed long segment myelitis extending from D1 to D4 thoracic segments and at D11-D12 (conus) and lesions in subcortical white matter of frontal and temporal lobes, dorsal pons, and right cerebellar peduncle (Figs. 2 3). She was treated with pulse steroids and rituximab. On follow-up after 19 weeks, her sensory symptoms and sphincter disturbances had resolved and EDSS had improved to 2 (Table 1).
|
Patient number |
1 |
2 |
3 |
4 |
5 |
6 |
Total cohort |
|---|---|---|---|---|---|---|---|
|
Abbreviations: ANA, antinuclear antibody; ARR, absolute risk reduction; b, bilateral; BS, brainstem; CoN, conus medullaris; CSF, cerebrospinal fluid; EDSS, expanded disability status scale; ESR, erythrocyte sedimentation rate; LETM, longitudinally extensive transverse myelitis; Lt, left; M, motor; MOG, myelin oligodendrocyte glycoprotein; MRI, magnetic resonance imaging; N/A, not available; Neg, negative; OCB, oligoclonal bands; ON, optic neuritis; P100, prolonged P100 latency; Pos, positive; PV, periventricular; rt, right; RTX, rituximab; S, sensory; SC, subcortex; SS-B-anti, Sjögren's syndrome type-B antibody; TM, transverse myelitis; UR, urologic; V, visual; VEP, visual evoked potential. a mean value. |
|||||||
|
Age |
29 |
35 |
39 |
25 |
57 |
36 |
36.8a |
|
Gender |
F |
M |
F |
M |
M |
F |
1:1 (M:F) |
|
Onset phenotype |
TM |
ON (bl) |
ON (lt) |
ON (rt) |
ON (rt) |
TM |
– |
|
Symptom at onset |
UR |
V |
V |
V |
V |
M,S,UR |
– |
|
Pyramidal signs |
Yes |
Yes |
No |
No |
Yes |
Yes |
66.70% |
|
EDSS at onset |
4 |
4 |
1 |
3 |
3 |
6 |
3.5a |
|
No. of attacks |
1 |
3 |
2 |
4 |
1 |
1 |
2a |
|
Relapse phenotype |
Nil |
ON (b) |
ON (lt) |
ON (rt) |
Nil |
Nil |
– |
|
ARR (before treatment) |
0 |
0.32 |
1.5 |
2.77 |
0 |
0 |
0.77a |
|
ESR (mm/1st h) |
9 |
2 |
20 |
2 |
7 |
18 |
9.7a |
|
CSF cells (cells/mm3) |
71 |
Nil |
Nil |
Nil |
17.8a |
||
|
CSF protein (mg/dL) |
17 |
N/A |
86 |
11 |
N/A |
36 |
37.5a |
|
CSF OCB |
Pos |
Neg |
Neg |
Neg |
25% |
||
|
Other autoimmunity |
Nil |
Nil |
ANA anti-SSB |
Nil |
Nil |
Nil |
16.70% |
|
MRI lesions |
LETM, CoN |
ON |
SC |
PV |
PV |
SC,BS, LETM, CoN |
– |
|
VEP in asymptomatic eye |
Nil |
Absent B/L |
Normal |
P100 |
P100 |
N/A |
– |
|
Immunotherapy |
RTX |
RTX |
RTX |
RTX |
RTX |
RTX |
– |
|
Relapse after therapy |
Nil |
Nil |
Nil |
Nil |
Nil |
Nil |
– |
|
Follow-up duration (mo) |
8 |
7 |
6 |
3 |
5 |
5 |
5.7a |
|
Disease duration (mo) |
8 |
82 |
14 |
16 |
59 |
5 |
30.7a |
|
Prednisolone dose at follow-up (mg/d) |
10 |
10 |
0 |
0 |
0 |
0 |
– |
|
EDSS at follow-up |
2 |
3 |
1 |
1 |
3 |
2 |
2a |

-
Fig. 2 Sagittal T2 MRI showing hyperintense signal in D1 to D4 thoracic segments and conus medullaris (arrows). MRI, magnetic resonance imaging.
Fig. 2 Sagittal T2 MRI showing hyperintense signal in D1 to D4 thoracic segments and conus medullaris (arrows). MRI, magnetic resonance imaging.
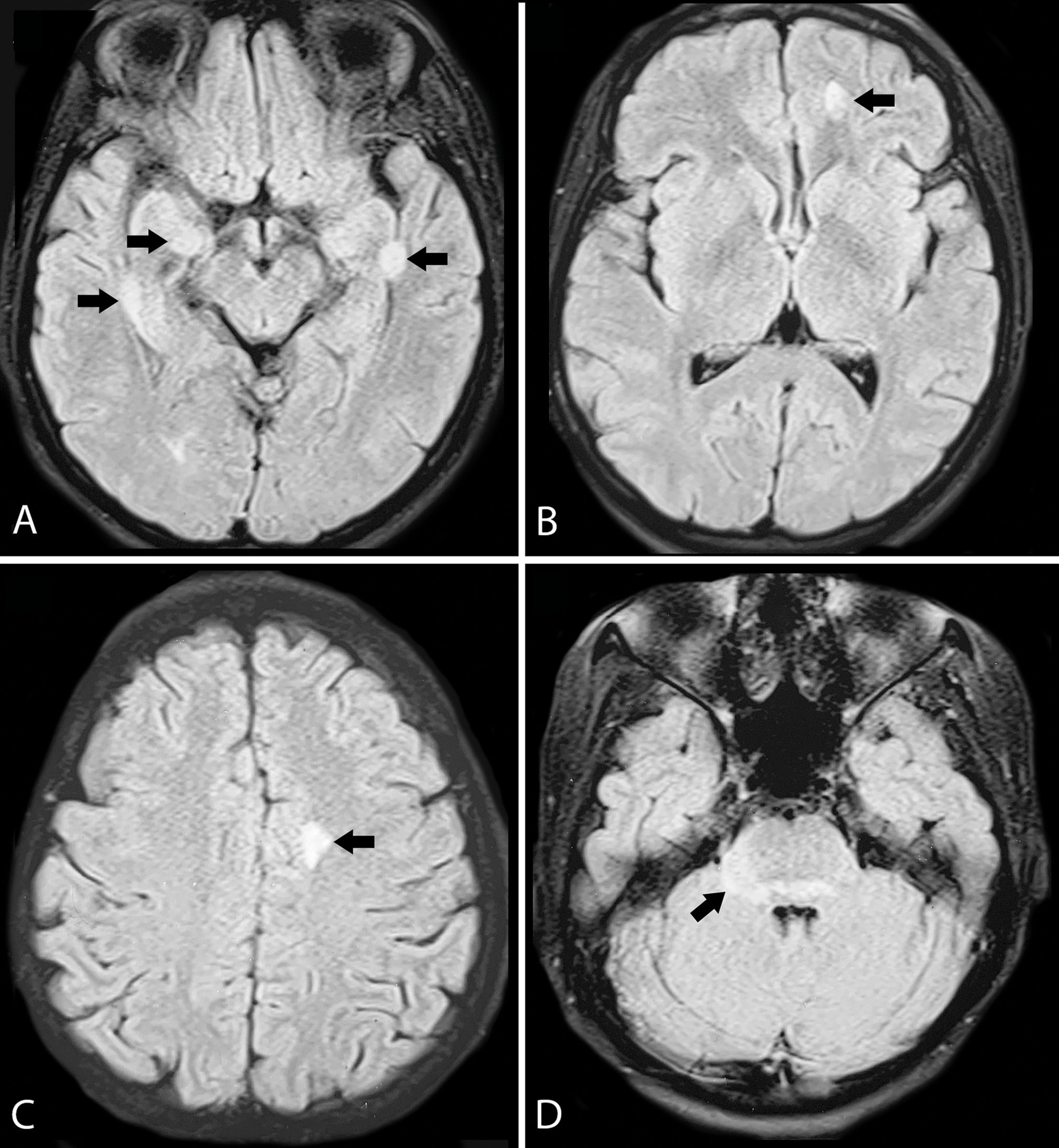
-
Fig. 3 Axial T2 FLAIR MRI showing (A) hyperintense lesions in subcortical white matter of both temporal lobes (arrows); (B,C) Subcortical white matter of left frontal lobe (arrows); (D) Dorsal pons and right cerebellar peduncle (arrows). FLAIR, fluid-attenuated inversion recovery; MRI, magnetic resonance imaging.
Fig. 3 Axial T2 FLAIR MRI showing (A) hyperintense lesions in subcortical white matter of both temporal lobes (arrows); (B,C) Subcortical white matter of left frontal lobe (arrows); (D) Dorsal pons and right cerebellar peduncle (arrows). FLAIR, fluid-attenuated inversion recovery; MRI, magnetic resonance imaging.
Discussion
There are only limited reports of MOG antibody disease from India.7 8 In our study, MOG antibodies were seen in 6 out of the 47 (12.8%) cases of CNS demyelination admitted to our institute in a span of 8 months. None of the patients were double positive for MOG and AQP-4 antibodies. The mean follow-up period was 5.7 months (range 3–8 months). The mean age was 37 years (range 25–57 years), which is in par with international data.9 The male: female ratio was 1:1, in sharp contrast to AQP-4 positive NMO which has a strong female preponderance (male: female = 1:9).10
Optic neuritis seen in four out of six patients (66.7%) was the most common presentation in our study, among which three (75%) had bilateral optic neuritis (either symptomatic or subclinical as evidenced by prolonged P100 latency in the unaffected eye) and is typical of MOG antibody disease.11 Patient 4 who had two past attacks of bilateral optic neuritis, presented with visual symptoms only in the left eye, probably because the visual recovery from past attacks was poor in the right eye.
Long segment transverse myelitis was the presentation in two patients (33.3%). Urological symptoms due to involvement of conus medullaris were seen in both the cases and are thought to be a characteristic feature of MOG antibody disease.12
The disease was relapsing in three out of six (50%) cases and all the relapses occurred in the optic nerve. The median time between relapses was 6 months, and relapse as late as 16 years after the first attack was seen in one patient, indicating the disease may have a chronic course. Even though patient 4, had a history of progressive unilateral visual loss without clear relapses, it might be due to recurrent and frequent episodes of optic neuritis in the same eye with poor visual recovery in between, mimicking a progressive disease. An interesting feature was that relapses occurred in the same eye as that of the first attack, and in the case of symptomatic bilateral optic neuritis, all relapses were bilateral. Patients with transverse myelitis had no relapses, probably because they were given immunotherapy after the first attack.
CSF pleocytosis was found in one patient (16.7%) and has been described in about half of the patients with MOG antibody disease.9 Elevated CSF protein without pleocytosis was seen in one patient and is seen in approximately 10% of the cases. CSF OCB was found in one patient and can occur in up to 15% of the patients.13 Coexisting autoimmune disease is uncommon in MOG as opposed to NMO and was seen in only one patient in our series, who had positive ANA and anti-SSB, although no sicca symptoms or any other systemic features of a connective tissue disorder was present.
The classical MRI lesions seen in MOG antibody disease include longitudinally extensive transverse myelitis with involvement of conus medullaris, deep gray matter involvement, large bilateral asymmetric white matter lesions, extensive bilateral anterior optic nerve involvement with swelling of intraorbital segment, and fluffy brainstem lesions in the pons, medullar, or cerebellar peduncles.12 Even though both our cases of transverse myelitis had lesions in the conus medullaris, the brain MRI findings in our series were not classical of MOG-antibody disease.
MOG-antibody disease is known to relapse in approximately 50% cases and most relapses tend to cluster after the initial attack. A variety of immunomodulators like intravenous immunoglobulin, rituximab, azathioprine, mycophenolate mofetil, cyclophosphamide, and cyclosporine have been tried to prevent relapses in MOG-antibody disease, and current therapies are center specific with no consensus or guidelines. In our experience with rituximab (two had maintenance prednisolone at 10 mg/d), all patients maintained remission without any relapses and EDSS improved in four out of five patients under follow-up (with the score remaining static without disability progression in the other two patients). None of the patients had any treatment-related side effects.
An important limitation of our study is that it did not include any pediatric cases. MOG antibody disease is seen in up to 40% cases of ADEM, especially in relapsing ADEM and ADEM-optic neuritis.
To conclude, MOG-antibody disease should be suspected in all the cases of NMO-SD who present with bilateral optic neuritis, longitudinally extensive myelitis with conus involvement, and urogenital symptoms. The disease tends to relapse early after the first attack and most relapses occur in the optic nerve. Rituximab therapy is safe, well-tolerated, and effective in preventing relapses and preventing disability progression.
Conflict of Interest
None declared.
References
- Differential ultrastructural localization of myelin basic protein, myelin/oligodendroglial glycoprotein, and 2′,3′-cyclic nucleotide 3′-phosphodiesterase in the CNS of adult rats. J Neurochem. 1989;52(1):296-304.
- [Google Scholar]
- The structure and function of myelin oligodendrocyte glycoprotein. J Neurochem. 1999;72(1):1-9.
- [Google Scholar]
- T- and B-cell responses to myelin oligodendrocyte glycoprotein in experimental autoimmune encephalomyelitis and multiple sclerosis. Glia. 2001;36(2):220-234.
- [Google Scholar]
- Myelin oligodendrocyte glycoprotein: deciphering a target in inflammatory demyelinating diseases. Front Immunol. 2017;8:529.
- [Google Scholar]
- Neuromyelitis optica spectrum disorders with aquaporin-4 and myelin-oligodendrocyte glycoprotein antibodies: a comparative study. JAMA Neurol. 2014;71(3):276-283.
- [Google Scholar]
- MOG cell-based assay detects non-MS patients with inflammatory neurologic disease. Neurol Neuroimmunol Neuroinflamm. 2015;2(3):e89.
- [Google Scholar]
- Anti myelin oligodendrocyte glycoprotein associated immunoglobulin G (AntiMOG-IgG)-associated neuromyelitis optica spectrum disorder with persistent disease activity and residual cognitive impairment. Ann Indian Acad Neurol. 2017;20(4):411-413.
- [Google Scholar]
- Serological markers associated with neuromyelitis optica spectrum disorders in South India. Ann Indian Acad Neurol. 2016;19(4):505-509.
- [Google Scholar]
- Clinical presentation and prognosis in MOG-antibody disease: a UK study. Brain. 2017;140(12):3128-3138.
- [Google Scholar]
- Epidemiology of neuromyelitis optica in the United States: a multicenter analysis. Arch Neurol. 2012;69(9):1176-1180.
- [Google Scholar]
- Optic neuritis in patients with anti-MOG antibodies spectrum disorder: MRI and clinical features from a large multicentric cohort in France. J Neurol. 2017;264(10):2173-2175.
- [Google Scholar]
- Myelin oligodendrocyte glycoprotein (MOG) antibody-associated disease: practical considerations. Pract Neurol. 2019;19(3):187-195.
- [Google Scholar]
- OFSEP and NOMADMUS Study Group. Clinical spectrum and prognostic value of CNS MOG autoimmunity in adults: the MOGADOR study. Neurology. 2018;90(21):e1858-e1869.
- [Google Scholar]




