
Translate this page into:
Clinical and Genetic Characteristics of Leukodystrophies in Africa
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Recent understanding of the genetic basis of neurological disorders in Africa has grown rapidly in the last two decades. Africa harbors the largest genetic repertoire in the world which gives unique opportunity to discover novel variant, genes, and molecular pathways associated with various neurological diseases. Despite that, large-scale genomic and exome studies are severely lacking especially for neglected diseases such as leukodystrophies. This review aims to shed light on the currently developed research in leukodystrophies in Africa. We reviewed all research articles related to “Leukodystrophy in Africa” published in Medline/PubMed and Google Scholar databases up to date. We found very few studies in leukodystrophy from Africa, especially from the Sub-Saharan regions. Metachromatic leukodystrophy was the most studied type of leukodystrophy. Published studies from North Africa (Tunisia, Morocco, and Egypt) were very limited in either sample size (case studies or single/few family studies) or molecular methods (targeted sequencing or polymerase chain reaction-restriction fragment length polymorphisms). More studies (GWAS or large family studies) with advanced techniques such as exome or whole genome sequencing are needed to unveil the genetic basis of leukodystrophy in Africa. Unmasking novel genes and molecular pathways of leukodystrophies invariably lead to better detection and treatment for both Africans and worldwide populations.
Keywords
Africa
genetics
leukodystrophy
review

INTRODUCTION
Leukodystrophies are a group of heritable metabolic disorders resulting from defective myelination of the central nervous system. The diseases present commonly in the first few months of life with hypotonia turning gradually into spasticity (spastic diplegia or quadriplegia). The spastic limbs severely impair purposeful movement which makes the disease very irritating to the parents, especially when associated with developmental regression, dyskinesia, ataxia, or seizures. In the final stages of the disease developing after a variable period of time, the child can no longer recognize his/her parents has difficulty in swallowing and breathing. Death is invariable in most cases.[1]
There are currently dozens known different types of leukodystrophies. Each with its own genetics and clinical manifestations but they all share white matter signals on brain MRI. The most common and studied forms of leukodystrophies are Metachromatic leukodystrophy (MLD), Krabbe disease, Canavan disease, Alexander disease, and X-linked adrenoleukodystrophy (X-ALD).
Epidemiological data on leukodystrophies are scarce even in the developed countries. Almost a dozen different types of leukodystrophies have still unknown genetic causation.[2] The situation is much more aggravated in Africa, where limited resources and lack of expertise stand against advanced research development. On the other hand, the associated high burden of neurological disorders with the currently well-known genomic heterogeneity of African populations[34] allows for a good opportunity to discover novel variants, genes, and molecular pathways that underlie the pathogenesis of most neurological disorders.[5] This review aimed to shed light on the currently developed research in leukodystrophies in Africa, highlighting the significant findings and gaps in knowledge for the hope of increasing the awareness of researchers, clinicians, and global organizations for participating in the research of leukodystrophies. Unmasking novel genes and molecular pathways of leukodystrophies will invariably lead to better detection and treatment for both Africans and worldwide populations.
METHODS
In this review, we followed the guidelines developed by the Preferred Reporting Items for Systematic Reviews and Meta-Analyses groups. The electronic databases Medline/PubMed and Google Scholar (1946 to Nov 2016) were searched to identify studies published up to date. The search strategy consisted of free-text words and subject headings related to “Leukodystrophy/leukodystrophies/leucodystrophy/leucodystrophies AND Africa” were added. In addition, articles related to “leukodystrophy” AND specific African countries (Tunisia, Egypt, Algeria, Morocco, Libya, Ethiopia, Eritrea, South Africa, Sudan, Nigeria, and Ghana) were also searched for. All different types of studies were included, and none was excluded on the basis of quality.
RESULTS
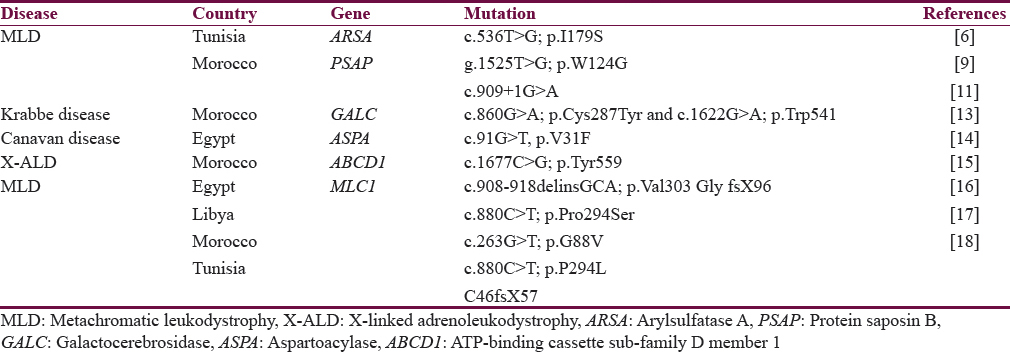
Studies in leukodystrophy in Africa are very geographically uneven. Most studies were done in North Africa (Tunisia and Morocco) with very few studies from Sub-Saharan regions. Published studies from North Africa were very limited in either sample size (case reports or single/few multiple family studies) or molecular methods (targeted sequencing or polymerase chain reaction-restriction fragment length polymorphisms) both preclude against making generalizations. Almost no study involved high throughput techniques such as whole exome/genome sequencing or microarray. Most types of studied leukodystrophies were MLD,[6789101112] Krabbe disease,[13] Canavan disease,[14] X-ALD,[15] and megalencephalic leukoencephalopathy.[161718] The results are summarized in Table 1.
DISCUSSION
Metachromatic leukodystrophy
MLD is a type of leukodystrophy caused by deficiency in the enzyme arylsulfatase A (ARSA), which is responsible for breaking down sulfatide compounds in neuronal myelin. The deficiency leads to accumulation of sulfatides in the brain, peripheral nerves, and other extraneural tissues. The disease manifests as progressive spasticity, optic atrophy, seizures, peripheral neuropathy, and intellectual impairment. MLD has three major types: late infantile-, juvenile-, and adult-onset types; ordered from worst to better prognosis. Few MLD patients with normal ARSA level have deficiency in the activator protein saposin B (PSAP) gene.[19] The disease is untreatable, but some success cases were successfully treated using enzyme replacement, bone marrow transplantation, or gene therapy.[20]
The prevalence of MLD in Africa is unknown. However, epidemiologic reports suggest that the disease is not uncommon. In a study by Barboura et al., 2.5% of Tunisian patients who present with progressive mental and behavioral deterioration were found to have MLD, distributed almost equally among the three different types.[7] However, in this study, only MLD was investigated, overlooking the other types of leukodystrophy which makes the true prevalence of leukodystrophies really underestimated.
Mutations in ARSA gene causing MLD were reported worldwide, but the most common are (c.459+1G>A, c.1204+1G>A, p. Pro426Leu, and p. Ile179Ser) mutations. The latter mutation was found in a 44-year-old woman diagnosed with adult type MLD from Tunisia.[6] Novel mutations were also found: A T→G substitution, g.1525 T>G; p.W124G in exon 2 of ARSA gene was detected in two Tunisian patients with MLD.[9] However, the limited sampled patients in these studies make it difficult to infer the spectrum of mutations in leukodystrophy in Tunisian population.
Mutations in the less common PSAP gene were also reported from Africa. In Morocco, one family diagnosed with MLD with normal ARSA enzyme levels had a homozygous splicing mutation (c.909+1G>A) in the PSAP gene causing loss of the entire exon 8.[11]
Measurement of ARSA enzyme level is a poor predictor for MLD since some healthy individuals were discovered to have very low enzyme levels. These patients have variations in the normal ARSA gene termed the pseudodeficiency allele (ARSA-PD). In Tunisia, the prevalence of the pseudodeficiency allele in the general population was found to be common (17.4% for the overall sample), with c.1049A (G and c.*96A G) frequencies of 25.6% and 17.4%, respectively.[8] Very rarely patients may present with both the (ARSA-MLD) and (ARSA-PD) alleles as has been found in one patient from Tunisia.[12]
Krabbe disease
Krabbe disease is a progressive metabolic disorder with 90% mortality rate before age 2 years in its late infantile form. It is one of the worst known types of leukodystrophy. Children appear normal at birth, but few months later, they develop extreme irritability, spasticity, and developmental regression. The disease is a classic example for an autosomal recessive disease with multispecies involvement, not only humans but monkeys, mice, and dogs also can be affected.[21] Almost all patients have deficiency in galactocerebrosidase (GALC) enzyme which results from a 30-kb deletion in the GALC gene occurring in 80% of patients from European and Mexican origins.[22] The first (and only) reported case of Krabbe disease from Africa was from Morocco in an 11-month-old male presented with symptoms of Krabbe disease and low level of GALC enzyme. The patient had a novel heterozygous compound mutation in GALC gene. The two mutations were c.860G >A; p. Cys287Tyr and c.1622G >A; p. Trp541.[13] Novel mutations in GALC gene continue to be discovered worldwide, and recently a c.943delG mutation was identified in an infant of a Turkish family with Krabbe disease with enlargement of both optic nerves.[23]
Canavan disease
Canavan disease is characterized by hypotonia, macrocephaly, and poor development. Gradually, hypotonia turns into spasticity leading to difficulty in speech, sitting, and ambulation. The disease is more common in Ashkenazi Jewish and results from mutations that differ from other worldwide populations. Aspartoacylase (ASPA) gene is the only gene currently associated with Canavan disease. The gene encodes an enzyme that catabolizes N-acetylaspartic acid (NAA); however, the pathophysiology which leads the accumulation of NAA to cause neuronal demyelination is still debated.[24] Three pathogenic variants (p. Glu285Ala, p. Tyr231Ter, and p. Ala305Glu) cause the majority of cases of Canavan disease in the world.[25] None of these mutations were reported from Africa. However, a novel homozygous missense variant (c.91G>T; p.V31F) in the ASPA gene however was found in a 2-year-old child with severe Canavan disease from Egypt.[14]
X-linked adrenoleukodystrophy
X-ALD is a form of leukodystrophy that affects both the nervous system and the adrenal cortex. Patients (most commonly boys) present with cognitive impairment, behavioral abnormality, and motor dysfunction. Almost all cases are caused by mutations in the ATP-binding cassette sub-family D member 1 (ABCD1) gene which leads to accumulation of very long chain fatty acids in different tissues leading to a complex clinical presentation.[26] Pathogenic variants reported worldwide were most commonly deletion/duplication. However, a missense mutation (c.1677C>G; p. Tyr559) in exon 7 of the ABCD1 gene was reported in a child from Morocco.[15]
Megalencephalic leukoencephalopathy with subcortical cysts
MLC with subcortical cysts is a rare form of leukodystrophy characterized by mild developmental delay, seizures, ataxia, and early onset macrocephaly. The disease is caused by mutations in commonly the megalencephalic leukoencephalopathy (MLC1) gene (MLC for MLC with subcortical Cysts). Defects in this gene lead to a disorder in water/ion hemostasis in brain astrocytes leading to white matter edema.[27] Known mutations are most commonly homozygous missense or compound heterozygous and few reported insertions/deletions.[28] Different types of mutations in MLC1 gene were found in one study from Egypt: deletion/insertion in exon 11 (c.908–918delinsGCA, p. Val303 Gly fsX96) in two families and a missense mutation in exon 10 (c.880 C >T; p. Pro294Ser) was identified in one family.[16] c.263G >T; p.G88V and c.880C >T; p.P294 L MLC1 gene mutations were also found in patients with Libyan and Moroccan origins, respectively.[17] A novel homozygous C46fsX57 mutation was found in a patient from Tunisia.[18] More interestingly, patients with severe muscle–eye–brain disease-like phenotype from Libya with missense mutation c.2006G>T; p. Cys669Phe in the β-subunit of dystroglycan gene, presented with MRI findings similar to that of megalencephalic leukoencephalopathy.[10] Recently discovered mutations in another gene HEPACAM were also associated with MLC.[27] Implication of this gene in MLC in patients from Africa is not yet found.
Other leukodystrophies
Pelizaeus–Merzbacher-like disease is a rare form of hypomyelinating leukodystrophy caused by mutations in Gap junction gamma-2 gene.[29] Mutations in the promoter region (c.-167A>G) were found in 4 Tunisian families with this disease.[30]
CONCLUSION
All currently published articles in leukodystrophy from Africa are limited and can barely give a true picture about the true burden of the disease and its associated genetic component, especially for a continent with the largest genetic repertoire in the world. The true genetic basis of inherited leukodystrophies in Africa is yet to be found.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1993. Leukodystrophy Overview, GeneReviews®. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24501781
- Childhood leukodystrophies: A clinical perspective. Expert Rev Neurother. 2011;11:1485-96.
- [Google Scholar]
- The genetic structure and history of Africans and African Americans. Science. 2009;324:1035-44.
- [Google Scholar]
- The African genome variation project shapes medical genetics in Africa. Nature. 2015;517:327-32.
- [Google Scholar]
- Neurogenomics in Africa: Perspectives, progress, possibilities and priorities. J Neurol Sci. 2016;366:213-23.
- [Google Scholar]
- Diagnostic strategy of metachromatic leukodystrophy in Tunisia. Ann Biol Clin (Paris). 2011;69:465-9.
- [Google Scholar]
- Brain MRI and biological diagnosis in five Tunisians MLD patients. Diagn Pathol. 2012;7:11.
- [Google Scholar]
- Determination of arylsulfatase a pseudodeficiency allele and haplotype frequency in the Tunisian population. Neurol Sci. 2016;37:403-9.
- [Google Scholar]
- Identification of a new arylsulfatase A (ARSA) gene mutation in Tunisian patients with metachromatic leukodystrophy (MLD) J Neurol Sci. 2009;287:278-80.
- [Google Scholar]
- Homozygous dystroglycan mutation associated with a novel muscle-eye-brain disease-like phenotype with multicystic leucodystrophy. Neurogenetics. 2013;14:205-13.
- [Google Scholar]
- A novel homozygous splicing mutation in PSAP gene causes metachromatic leukodystrophy in two Moroccan brothers. Neurogenetics. 2014;15:101-6.
- [Google Scholar]
- An unusual homozygous arylsulfatase: A pseudodeficiency in a metachromatic leukodystrophy Tunisian patient. J Child Neurol. 2010;25:82-6.
- [Google Scholar]
- Clinical and molecular report of novel GALC mutations in Moroccan patient with Krabbe disease: Case report. BMC Pediatr. 2015;15:182.
- [Google Scholar]
- Novel mutation in an Egyptian patient with infantile Canavan disease. Metab Brain Dis. 2016;31:573-7.
- [Google Scholar]
- A novel mutation in the ABCD1 gene of a Moroccan patient with X-linked adrenoleukodystrophy: Case report. BMC Neurol. 2015;15:244.
- [Google Scholar]
- Clinical, neuroimaging, and genetic characteristics of megalencephalic leukoencephalopathy with subcortical cysts in Egyptian patients. Pediatr Neurol. 2014;50:140-8.
- [Google Scholar]
- Genetic heterogeneity of megalencephalic leukoencephalopathy and subcortical cysts. Neurology. 2003;61:534-7.
- [Google Scholar]
- Megalencephalic leukoencephalopathy with subcortical cysts in a Tunisian boy. J Child Neurol. 2009;24:87-9.
- [Google Scholar]
- 1993. Arylsulfatase A Deficiency, GeneReviews®. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20301309
- Metachromatic leukodystrophy: Disease spectrum and approaches for treatment. Best Pract Res Clin Endocrinol Metab. 2015;29:261-73.
- [Google Scholar]
- A new mutation in an infant with Krabbe disease accompanied by enlargement of the optic nerves. Acta Neurol Belg. 2017;117:319-321.
- [Google Scholar]
- Canavan disease: Clinical features and recent advances in research. Pediatr Int. 2014;56:477-83.
- [Google Scholar]
- Adrenoleukodystrophy-Neuroendocrine pathogenesis and redefinition of natural history. Nat Rev Endocrinol. 2016;12:606-15.
- [Google Scholar]
- Megalencephalic leukoencephalopathy with subcortical cysts: Chronic white matter oedema due to a defect in brain ion and water homoeostasis. Lancet Neurol. 2012;11:973-85.
- [Google Scholar]
- 1993. Megalencephalic Leukoencephalopathy with Subcortical Cysts, GeneReviews®. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20301707
- Expanded spectrum of Pelizaeus-Merzbacher-like disease: Literature revision and description of a novel GJC2 mutation in an unusually severe form. Eur J Hum Genet. 2013;21:34-9.
- [Google Scholar]
- Molecular confirmation of founder mutation c.-167A>G in Tunisian patients with PMLD disease. Gene. 2013;513:233-8.
- [Google Scholar]




