
Translate this page into:
Calf heads on a trophy sign: Miyoshi myopathy
Address for correspondence: Dr. M. Mundayadan Shyma, Department of Neurology, TD MCH, Alappuzha - 688 005, Kerala, India. E-mail: mm.shyma@gmail.com
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Miyoshi myopathy is an autosomal recessive distal myopathy with predominant involvement of the posterior calf muscles attributed to mutations in the dysferlin gene. We report a 26-year-old male, born of nonconsanginous parentage. He noticed weakness and atrophy of leg muscles with inability to walk on his heels. The creatine kinase concentration was high. The electromyography showed myopathic pattern and the muscle biopsy disclosed dystrophic changes with absence of dysferlin. Miyoshi myopathy may be distinct among the hereditary distal myopathies. There are only few reported cases of Miyoshi myopathy in the world literature. In India only 12 cases were reported who had classical features of Miyoshi myopathy. Our's is a typical case of Miyoshi myopathy, with an affected twin sister as well. He also had “calf heads on a trophy sign” on physical examination, which is considered to be pathognomonic of this disease.
Keywords
“Calf heads on trophy” sign
distal myopathy
familial miyoshi myopathy

Introduction
Myopathies usually present as weakness without atrophy involving primarily pelvic and scapular muscles.[12] However there is a group of rare myopathies with distinct characteristics that affect predominantly distal muscles. They can be identified by the following features: Autosomal pattern of genetic inheritance, early or late onset distal muscle atrophy, increased creatine kinase (CK) and abnormal muscle biopsy.[13] The Miyoshi myopathy may be distinct among the hereditary distal myopathies, first described in the medical literature in 1967 in Japan by Miyoshi et al.[24] We describe a family of distal myopathy where the diagnostic clue towards the underlying diagnosis was pointed out by a physical examination finding of “calf heads on a trophy” sign.
Case Report
A 26-year-old male presented to us with progressive muscle weakness and wasting of 3 years duration with inability to run or climb stairs. Disease progressed gradually to weakness, wasting of the hand and forearm muscles. There was no involvement of facial muscles, eye, oropharyngeal and neck muscles. There were no muscle cramps, aches, pains or fasciculations. He was born out of nonconsanginous marriage as first of the twins with normal development and no mental retardation. On examination he had increased lumbar lordosis without evidence of cataract. Folstein's Mini Mental Status Examination was normal (30/30). There was symmetrical wasting of trapezius, sternocleidomastoid, supraspinatus, forearm muscles, hand and leg muscles (distal >proximal) with winging of scapula associated with symmetrical weakness of hand and leg muscles (grade 4–4/5 power, distal >proximal). “Calf heads on a trophy sign” was present and Gowers’ sign was positive [Figures 1a–d and 2a and b]. The tendon reflexes were present and symmetric. Waddling gait with difficulty in walking on tip toes was observed without evidence of thickened nerves or myotonia. Similar features were observed in his twin sister as well [Figure 3]. Routine investigations were normal. Serum CK was - 1040 IU/L (normal 60–100 IU/L), lactate dehydrogenase - 747 IU/L (normal: 105–333 IU/L). The electromyography (EMG) showed a myopathic pattern with muscle biopsy taken from the vastus lateralis revealing dystrophic changes and Immunostaining showed evidence of normal staining pattern for dystrophin (1, 2, 3), sarcaglycans (a, b, g, d), dystroglycans and merosin, except for total absence of dysferlin.
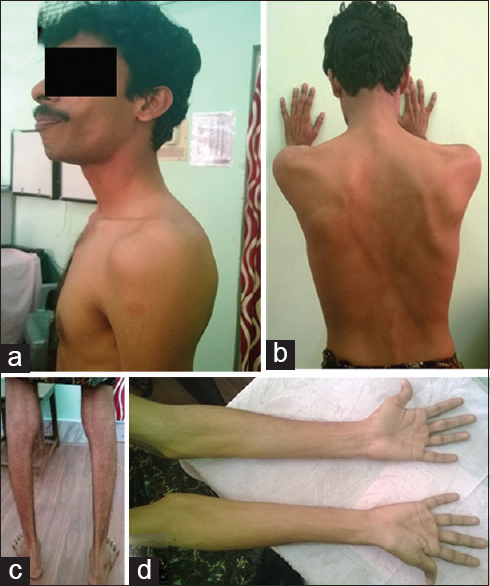
- (a) Deltoid sparing; (b) scapular winging; (c) wasting of calf muscles; (d) wasting of hand muscles
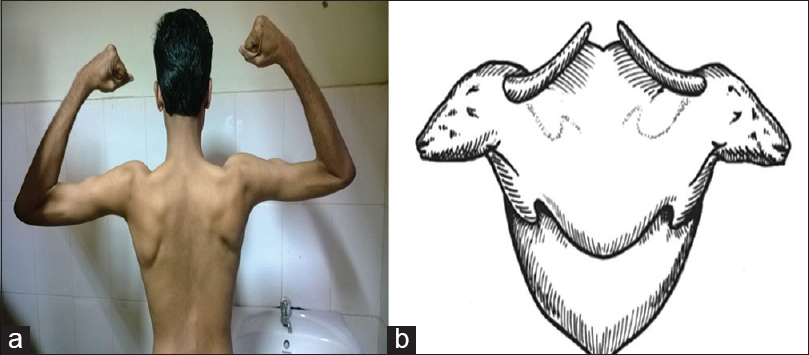
- Calf heads on a trophy sign. (a) Posture adopted to look for the calf heads on a trophy sign; (b) Diagrammatic representation matches with the sign shown in 2(a)
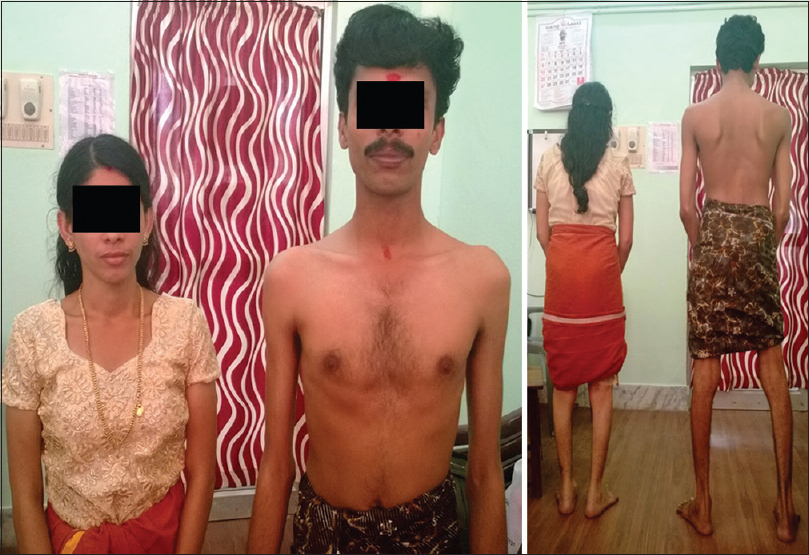
- Patient with his twin sister with similar clinical appearance
Discussion
Myopathies usually present proximal weakness with or without atrophy[12] involving mainly pelvic and scapular muscles. However, there is a group of rare myopathies with different characteristics that affect especially distal muscles. They can be identified by the following features: Autosomal dominant or autosomal recessive (AR) genetic inheritance, early or late onset distal muscle atrophy, increased CK and abnormal muscle biopsy findings.[13] The Miyoshi myopathy may be distinct among the hereditary distal myopathies. First described in the medical literature in 1967 in Japan by Miyoshi et al.[4]
Onset of the disease is in early adult life with weakness and atrophy of the leg muscles.[12] Recently genetic linkage to chromosome 2p12–14[1] has been established. It has an AR transmission. Difficulty in tiptoeing and climbing stairs is the initial symptom, with involvement of gastrocnemius and soleus muscles.[5] There is a slowly progressive weakness and the proximal muscles may be involved late.[25] “Calf heads on a trophy sign”[3] is a specific clinical sign that is almost specific for Miyoshy myopathy: First is a prominent deltoid muscle looking like a calf's heads with the mouth at the muscle's insertion and one of the several linear depressions appearing like a half-closed eye. Second is a cordlike upper border of the trapezius muscle appearing like a horn on the calf's heads. Third is a prominent upper medial part of the infraspinatus muscle with sharply demarcated wasting of the lower and lateral part, looking like a downward directed ear of the calf. Fourth is the spinous process of the scapula, which becomes prominent because of the wasted supraspinatus above it appearing like the upper border of the calf's neck. Fifth is the overall appearance of the upper back as that of a trophy. These “calf heads” appeared prominent over the upper back because of the hollowing of the interscapular region. The CK is usually high. The EMG shows a myopathic pattern and the muscle biopsy reveals dystrophic changes without the presence of rimmed vacuoles and absent dysferlin. It has been also described in USA, Italy, Spain, Germany, The Netherlands, Africa and Brazil. In Japan its incidence is approximately 1 in 440,000 inhabitants.[14]
The AR pattern of inheritance in combination with the clinical features, EMG, serum CK level and muscle biopsy in our patient were suggestive of Miyoshi myopathy. Muscle biopsy done in his twin sister was also suggestive of Miyoshi myopathy. In both of them the onset was in young age, between 20 and 23 years. The main symptom was distal lower limb weakness with difficulty to tiptoe. There was severe atrophy in gastrocnemius muscles.[5] This causes thin and tapering legs. This progression has been described in Miyoshi myopathy and it is the only distal myopathy that begins in the posterior part of the legs. The proximal lower limbs muscles may be involved after many years and the anterior tibial and the peroneal muscles are almost normal.[245] Our patient also showed “calf heads on trophy sign.”[3] The calf heads on a trophy sign was not visible in any patient with muscle disease other than Miyoshi myopathy and thus appears to be highly specific. As the final diagnosis nowadays is made with immunohistochemical staining of muscle biopsy specimens and genetic studies, the calf heads on a trophy sign may be useful in directing patients to appropriate tests, thus minimizing the time and cost of diagnosis. Recently it has been described that Miyoshi myopathy is due to the absence or decrease of a protein in the muscle membrane called dysferlin. This protein of 273k DA, made of 2080 amino acids might function in calcium mediated membrane fusion or trafficking.[56] The limb girdle muscular dystrophy 2B (LGMD 2B) has the same dysferlin locus like[6] Miyoshi myopathy. Meugfatt[7] et al. described a new method to differentiate Miyoshi myopathy from LGMD 2B. In Miyoshi myopathy there is a lack in blood dysferlin, as it exists in monocytes. The technique would permit a less invasive, faster and cheaper diagnosis and allows to quantify dysferlin levels when new treatments would be tested.[6] The high levels of CK in Miyoshi myopathy is due to great muscular lesion. Miyoshi et al. and Barohn[48] reported high level of this enzyme in asymptomatic patients’ relatives. Probably they were preclinical individuals or were heterozygous. Besides enzymatic increase, the EMG also confirms myopathic pattern with brief lasting potential, with small amplitude and polyphasic potentials. The immunocytochemistry on muscle biopsy performed in our patient showed absence of dysferlin in muscle membrane. Myopathic features with polyfocal regeneration and occasional necrosis-no inflammation and absence of rimmed vacuoles were observed in some fibres.[910] There are other distal myopathies that look like Miyoshi myopathy. They can have dominant or recessive autosomal patterns. The Nonaka's myopathy is a recessive autosomal disorder with the onset in young adults.[9] This myopathy shows weakness in feet dorsiflexor muscles and toe extensors (“fallen feet”). The CK is high and on muscle biopsy there are rimmed vacuoles, focal myofibrillar destruction and autophagocytoses.[101112] Welander's distal myopathy, Tibial muscular dystrophy (Udd's), Markesbery-Griggs distal myopathy, Laing's distal myopathy are all dominantly inherited late onset distal myopathies.[5910] Welander's myopathy involves wrist and finger extensors where as others have anterior tibial involvement.[5910] In our case two siblings were affected without clinical involvement of parents. Along with clinical features, high CPK level, myopathic EMG, the absence of dysferlin in muscle biopsy confirms the diagnosis of Miyoshi myopathy. We feel that in patients where the clinical possibility of a distal myopathy is strong, it is mandatory to perform genetic tests and muscle biopsy with immunohistochemical techniques, as is being stressed in our index case.
Conclusion
“Calf heads on a trophy sign” is not visible in any patient with muscle disease other than Miyoshi myopathy and thus appears to be highly specific. As the final diagnosis nowadays is made with immunohistochemical staining of muscle biopsy specimens and genetic studies, the calf heads on a trophy sign may be useful in directing patients to appropriate tests, thus minimizing the time and cost of diagnosis.
Source of Support: Nil.
Conflict of Interest: None declared.
References
- Linkage of Miyoshi myopathy (distal autosomal recessive muscular dystrophy) locus to chromosome 2p12-14. Neurology. 1995;45:768-72.
- [Google Scholar]
- Miyoshi-type distal muscular dystrophy. Clinical spectrum in 24 Dutch patients. Brain. 1997;120(Pt 11):1989-96.
- [Google Scholar]
- Autosomal recessive distal muscular dystrophy as a new type of progressive muscular dystrophy. Seventeen cases in eight families including an autopsied case. Brain. 1986;109(Pt 1):31-54.
- [Google Scholar]
- Distal myopathies. In: Engel AG, Franzini-Armstrong C, eds. Myology. New York: McGraw Hill; 1994. p. :1246-57.
- [Google Scholar]
- Identical dysferlin mutation in limb-girdle muscular dystrophy type 2B and distal myopathy. Neurology. 2000;55:1931-3.
- [Google Scholar]
- A novel blood based assay for limb girdle muscular dystrophy 2B and Miyoshi myopathy. Ann Neurol. 2002;51:129-33.
- [Google Scholar]
- Dysferlinopathy: A clinical and histopathological study of 28 patients from India. Neurol India. 2008;56:379-85.
- [Google Scholar]




