
Translate this page into:
Hashimoto's Encephalopathy Masquerading as Rapidly Progressive Dementia and Extrapyramidal Failure

Yasser Aladdin, MD, FRCPC Department of Neurology, King Abdulaziz Medical City/King Saud bin Abdulaziz University for Health Sciences P.O. Box: 12723, Jeddah 21483 Saudi Arabia yaladdin@ymail.com
This article was originally published by Thieme Medical and Scientific Publishers Pvt. Ltd. and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Abstract
Hashimoto's encephalopathy is a rare immune-mediated disorder characterized by subacute encephalopathy with elevated thyroid antibodies. Hashimoto's encephalopathy is also known as steroid-responsive encephalopathy associated with autoimmune thyroiditis. We report a rare presentation of Hashimoto's encephalopathy presenting with acute neuropsychiatric disturbances, rapidly progressive dementia, seizures, and extrapyramidal failure. Neuroimaging revealed multifocal vasculitides of major cerebral vessels that support the autoimmune vasculitic theory as the underlying pathogenesis for Hashimoto's encephalopathy. Unfortunately, permanent irreversible cerebral damage has already ensued before her presentation to our center, which rendered steroid therapy ineffective. Serological testing for Hashimoto's thyroiditis must be in the investigation of all rapidly progressive dementias as early diagnosis and timely management of autoimmune thyroiditis may salvage sizable and eloquent cerebral tissues. The rarity of the condition should not preclude the investigation of Hashimoto's disease even in the presence of normal levels of thyroid hormones. Delayed diagnosis may result in irreversibly catastrophic encephalopathy in patients who once presented with potentially curable dementia.
Keywords
Hashimoto's encephalopathy
rapidly progressive dementia
extrapyramidal failure
Saudi Arabia

Introduction
Hashimoto's encephalopathy, also known as steroid-responsive encephalopathy associated with autoimmune thyroiditis, is a rare immune-mediated disorder characterized by subacute encephalopathy with elevated thyroid antibodies and responsiveness to immunotherapy in the absence of other autoantibodies.1 Additional clinical features include seizures, psychiatric symptoms, myoclonus, tremor, stroke-like episodes, sleep abnormalities, and gait difficulties.2 Extrapyramidal failure with parkinsonism and choreoathetotic movements in Hashimoto's encephalopathy were rarely reported in the literature.3 The pathogenesis of Hashimoto's encephalopathy is not well elucidated despite several proposed hypotheses including a dysregulated immune system, cerebral vasculitis, recurrent demyelination, and neurotoxic effects of thyrotropin-releasing hormone.4 The autoimmune mechanism is the most plausible mechanism supported by the prompt and favorable clinical response to steroids that is paralleled by the serological response of lowered antithyroglobulin titers.5 In this study, we report a case of Hashimoto's encephalopathy presenting as neuropsychiatric disturbance, rapidly progressive dementia, extrapyramidal failure, and seizures mimicking spongiform encephalopathy.
Case Report
A 47-year-old female patient presented to the emergency room with a history of progressive decline in recent memory manifesting as forgetfulness of verbal and non-verbal events. Her symptoms started insidiously 2 years prior to her presentation as emotional lability with a tendency for crying associated with forgetfulness manifesting as misplacing items and misnaming individuals. She precipitously deteriorated as she developed insomnia with agitation and a relentless decline in memory. Moreover, her mobility declined with difficulty to initiate and sustain purposeful movements that rapidly and significantly advanced to impose on basic activities of daily living. She was assessed in another hospital, but her symptoms progressed over the last year as she became bedbound. Her ability to speak deteriorated in conjunction with her progression including forgetting words and failure to recall names that progressed into incoherent and incomprehensible sounds. She also developed generalized tonic–clonic seizures post-ictal confusion and sleepiness. Her past medical history was unremarkable apart from type 2 diabetes mellitus. Her family history is non-contributory for neurological conditions.
The neurological assessment revealed stable vital functions with equally reactive pupils and a full range of ocular motility. The patient was not following commands with global rigidity, bradykinesia, masked face, and anarthria. Deep tendon reflexes were brisk without clonus. Intermittent choreaoathetotic movements were noted in the upper limbs.
Full blood biochemical investigation was unremarkable apart from elevated thyroid peroxidase antibody at 97.72 (normal 1–16). Her thyroid stimulating hormone level was 1.19 mIU/L (normal range: 0.6–5 mIU/L), free T4 was 12.80 pmol/L (normal range: 9–19 pmol/L), and vitamin B12 was 593 pmol/L (normal range: 131–507 pmol/L). Brain computed tomography (CT) showed no acute intracranial insult, and carotid CT angiography showed long segments of narrowing and wall irregularities involving the left A2–A3 and inferior division of the left middle cerebral artery at the M2 segment. MRI of the brain showed multiple areas of stenosis at the ACA, MCA, and PCA that could be related to vasculitis. In addition, striking bilateral medial temporal lobe atrophy was observed (Fig. 1). Brain single-photon emission computed tomography (SPECT) scan showed a focal area of perfusion defect in both frontal and parieto-occipital lobes, particularly on the left side. In addition, the medial temporal lobes demonstrated a perfusion defect in correspondence of the bilateral temporal lobe atrophy (Fig. 2). EEG showed moderate slowing cerebral activity with no evidence of epileptiform discharges. Chest, abdomen, and pelvis imaging with CT was unremarkable to her condition. Thyroid ultrasound showed left lobe heterogeneous nodule measuring 1.1 × 1.1 × 1.3 cm with peripheral vascularity classified as TI-RADS 3, which is a highly probable benign nodule but requiring fine-needle aspiration (Fig. 3). Left thyroid fine-needle aspiration under ultrasound guidance showed benign findings consistent with colloid nodule background lymphocytic/Hashimoto's thyroiditis. CSF analysis was negative including protein 14-3-3 among other markers and antibodies. Serum ceruloplasmin and serum copper were normal despite mildly elevated urinary 24-hour copper at 1.84 μmol/day. Genetic testing using whole-exome sequencing was negative for genetic abnormalities. Brain biopsy was declined by the family.
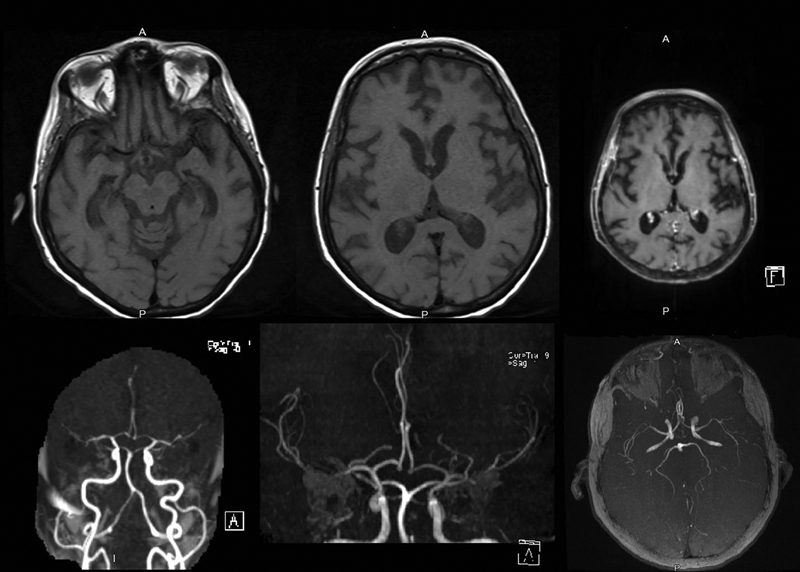
-
Fig. 1 MRI of the brain showing multiple areas of stenosis at the ACA, MCA, and PCA as well as striking bilateral medial temporal lobe atrophy.
Fig. 1 MRI of the brain showing multiple areas of stenosis at the ACA, MCA, and PCA as well as striking bilateral medial temporal lobe atrophy.
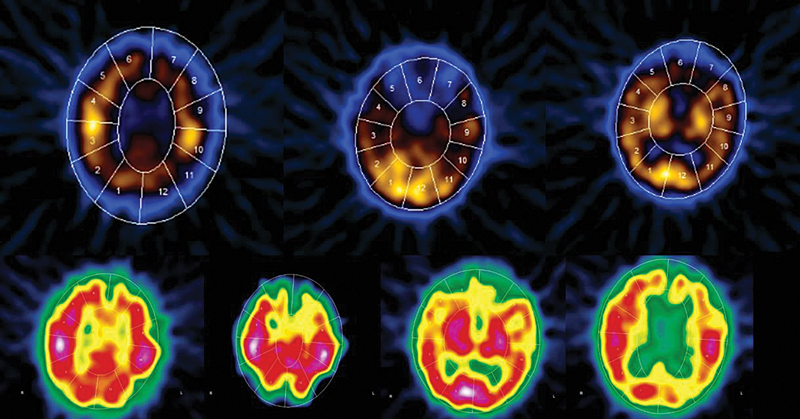
-
Fig. 2 SPECT scan showing a focal area of perfusion defect in the medial temporal lobes as well as both frontal and parieto-occipital lobes, particularly on the left side.
Fig. 2 SPECT scan showing a focal area of perfusion defect in the medial temporal lobes as well as both frontal and parieto-occipital lobes, particularly on the left side.
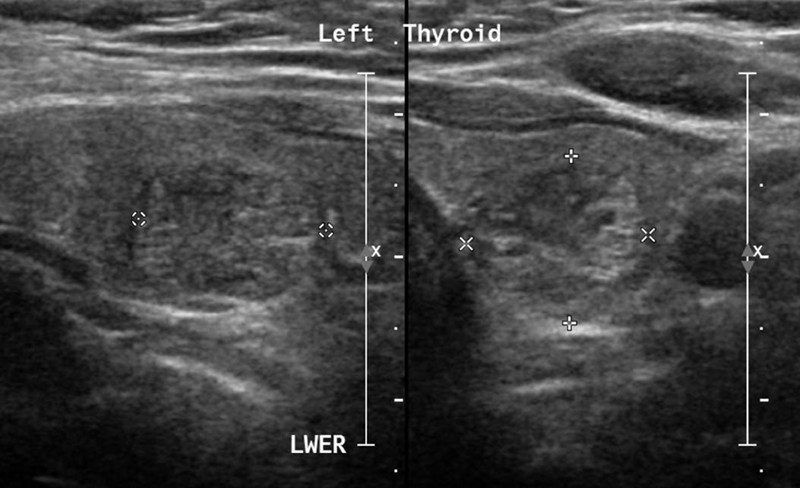
-
Fig. 3 Thyroid ultrasound showing left lobe heterogeneous nodule measuring 1.1 × 1.1 × 1.3 cm with peripheral vascularity classified as TI-RADS 3, which is a highly probable benign nodule but requiring fine-needle aspiration.
Fig. 3 Thyroid ultrasound showing left lobe heterogeneous nodule measuring 1.1 × 1.1 × 1.3 cm with peripheral vascularity classified as TI-RADS 3, which is a highly probable benign nodule but requiring fine-needle aspiration.
The patient was diagnosed with Hashimoto's encephalopathy and started on intravenous pulse steroid therapy (1000 mg methylprednisolone) with limited improvement. She was also started on levodopa/carbidopa, trihexyphenidyl, and rotigotine with remarkable, but transient improvement in speech and motor responsiveness. The course of pulse steroid therapy was repeated twice with limited response. In addition, a course of intravenous immunoglobulin was trialed without sensible changes in the clinical condition. The patient plateaued at severe rigidity and global bradykinesia with anarthria except for incomprehensible moaning along with total dependence on nursing care.
Discussion
Females are four to five times more likely to be affected by Hashimoto's encephalopathy, with a mean age of onset between 41 and 48 years, and our patient's demographics were in accordance with the published studies.6
The onset of Hashimoto's encephalopathy is usually acute or subacute and can present clinically with reversible dementia.7 Our patient presented with rapidly progressive dementia commencing with neuropsychiatric manifestations with subsequent evolution into seizures and extrapyramidal failure manifesting as a complex combination of global akinesia and choreoathetotic movements. The rapid disease progression was apparently paralleled by an expeditious pathology at the neuronal level resulting in severe caudate atrophy bilaterally with diffuse cerebral atrophy. This irreversible structural loss has resulted in irreversible dementia despite immunological treatment.
Cerebral manifestations of Hashimoto's disease are not necessarily the direct byproduct of hypothyroidism that occurs in end-stage Hashimoto's thyroiditis. Chronic autoimmune thyroiditis may encompass a long euthyroid period, which reflects that not merely the lack of thyroid hormones, but the impact of an autoimmune process in the pathogenesis of brain disorders in patients with Hashimoto's thyroiditis.8 Evidently, this was the case in our patient because she was euthyroid despite a tissue-proven diagnosis of Hashimoto's thyroiditis.
Diagnostic testing findings in Hashimoto's encephalopathy have been reported as non-specific with either normal MRI or non-specific signal abnormalities.9 Our patient demonstrated striking vascular abnormalities suggestive of vasculitis of large and medium arteries. Severe atrophy of the caudate and temporal lobes likely reflects a prolonged cerebral inflammation secondary to an occult long-standing Hashimoto's encephalopathy.
Current studies have suggested vasculitis as one potential pathogenesis of Hashimoto's encephalopathy.10 In a post-mortem study of a patient with Hashimoto's encephalopathy, lymphocytic infiltrates were observed within leptomeningeal veins and venules.11 CT angiography has also demonstrated vasculitis in patients with Hashimoto encephalopathy.12 The vasculitic theory was also proposed by Forchetti et al13 who interpreted the disruption of cerebral blood flow as a result of immune complex deposition of autoantibodies on cerebral microvasculature evidenced by diffuse and homogenous hypoperfusion on SPECT. The SPECT imaging in our patient showed even wider involvement of large and medium cerebral vessels resulting in multiple areas of perfusion defect in congruence with the previous report.
Nevertheless, Hashimoto's encephalopathy remains a diagnosis of exclusion. Toxic, infectious, metabolic, neoplastic, and other neuronal antibody syndromes should be excluded as was the case in our patient.14 The mysterious neuropsychiatric onset and precipitous evolution into seizures and a turbulent memory loss may mimic spongiform encephalopathy, particularly Creutzfeldt Jacob disease. The rarity of the condition should not justify skepticism for uncommon clinical conditions that include proper investigation for Hashimoto's disease. Delayed diagnosis may result in irreversible dementing illness and missing the diagnosis of one of the few potentially curable dementias.
The current standard treatment of Hashimoto's encephalopathy is corticosteroids and treatment of any concurrent thyroid disorder. Generally, the symptoms improve or completely resolve over a few months. In resistant cases, other immunotherapies may be used, including plasma exchange, IVIG, methotrexate, azathioprine, cyclophosphamide, and mycophenolate mofetil.15 In our case, we used IVIG and three courses of pulse steroid therapy with limited improvement. Other immunotherapies were not used due to her fragile clinical condition.
Hashimoto's encephalopathy may be under-diagnosed due to the versatile clinical presentations with protean clinical manifestations. Intuitive clinical acumen with a low index of suspicion could have salvaged many patients as the defining characteristic of Hashimoto's encephalopathy is the reversibility with immunosuppressive therapy. However, the lack of improvement in our patient might be attributed to the delay in presentation until permanent irreversible neurological damage. Hashimoto's encephalopathy should be on the shortlist of potentially curable dementias where early diagnosis and timely management may prevent permanent damage to critical structures within the central nervous system.
Conclusion
Hashimoto's encephalopathy may rarely present with a constellation of neuropsychiatric symptoms, rapidly progressive dementia, and extrapyramidal failure with atrophy of the caudate nucleus. Autoimmune mechanisms with vasculitic and non-vasculitic mechanisms are probably involved. The delayed diagnosis of Hashimoto's disease may have resulted in a catastrophic irreversible encephalopathy in a patient who presented with potentially curable dementia.
Conflict of Interest
None declared.
References
- Brain dysfunction and thyroid antibodies: autoimmune diagnosis and misdiagnosis. Brain Commun. 2021;3(2):a233.
- [Google Scholar]
- Hashimoto's encephalopathy presenting with progressive cerebellar ataxia. Neurosciences (Riyadh). 2019;24(4):315-319.
- [Google Scholar]
- Hashimoto's encephalopathy presenting with chorea. J Assoc Physicians India. 2015;63(9):83-84.
- [Google Scholar]
- The neuromythology of Hashimoto encephalopathy: The emperor has no clothes. Neurology. 2020;94(2):55-56.
- [Google Scholar]
- Hashimoto's encephalopathy presented with mutism: a case report. Gen Psychiatr. 2021;34(3):e100502.
- [Google Scholar]
- Hashimoto's encephalopathy: a brief review. Curr Neurol Neurosci Rep. 2014;14(9):476.
- [Google Scholar]
- Thyroid gland and brain: enigma of Hashimoto's encephalopathy. Best Pract Res Clin Endocrinol Metab. 2019;33(6):101364.
- [Google Scholar]
- MRI findings of two patients with Hashimoto encephalopathy. Cureus. 2021;13(6):e15697.
- [Google Scholar]
- Hashimoto's encephalopathy: postmortem findings after fatal status epilepticus. Neurology. 2003;61(8):1124-1126.
- [Google Scholar]
- Central nervous system vasculitis with positive antithyroid antibodies in an adolescent boy. Pediatr Neurol. 2011;45(3):189-192.
- [Google Scholar]
- Autoimmune thyroiditis and a rapidly progressive dementia: global hypoperfusion on SPECT scanning suggests a possible mechanism. Neurology. 1997;49(2):623-626.
- [Google Scholar]
- Hashimoto encephalopathy: a case report and a short revision of current literature. Acta Biomed. 2020;91(3):e2020087.
- [Google Scholar]
- Hashimoto encephalopathy: literature review. Acta Neurol Scand. 2017;135(3):285-290.
- [Google Scholar]




