
Translate this page into:
Neuromyelitis Optica Spectrum Disorders in Black African: Experience of Togo (2015–2020)
Kossivi Apetse, MD Service de Neurologie du CHU Campus, Faculté des Sciences de la Santé, Université de Lomé 01 BP: 1515, Lomé Togo kapetse@hotmail.com
This article was originally published by Thieme Medical and Scientific Publishers Pvt. Ltd. and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Abstract
Introduction Neuromyelitis optica spectrum disorders (NMOSD) would disproportionately affect blacks within mixed populations. However, they are rarely reported in black African. The objective of this work was to report the experience of Togo, a West African country in terms of NMOSD.
Methods This is a series of six cases diagnosed between 2015 and 2020 in the only three neurology departments in Togo. The diagnosis of NMOSD was made according to the criteria of the International Panel for NMO Diagnosis (2015) and the patients had a minimum clinical follow-up of 6 months after the diagnosis. The search for anti-aquaporin 4 (AQP4) antibodies was performed by immunofluorescence on transfected cells.
Results The mean age was 25.33 years and the sex ratio female/male was 5/1. The average time between the first attack and the diagnosis was 122.83 days. Clinically, there was isolated medullary involvement (2/6), simultaneous opticomedullary involvement (3/6), and area postrema syndrome (1/6). Five patients were anti-AQP4 positive. All six patients had extensive longitudinal myelitis. At 6 months of follow-up, there was one case of death and one case of blindness.
Conclusion The rarity of NMOSD cases in Togo could be linked to an underestimation. To better characterize the NMOSDs of the black African population, multicenter and multidisciplinary studies are necessary.
Keywords
anti-AQP4 antibody
neuromyelitis optica spectrum disorders
Togo
Africa

Introduction
Neuromyelitis optica (NMO) is a severe inflammatory demyelinating disease of the central nervous system (CNS) primarily affecting the optic nerves and spinal cord.1 Distinct from multiple sclerosis, it was long considered a monophasic opticomedullary disease.2 However, with the discovery of anti-aquaporin 4 autoantibodies (anti-AQP4), which are highly specific for the disease, the concept of the NMO spectrum disorders (NMOSD) appeared, which includes other clinical presentations, particularly encephalic, and clinical forms evolving in relapses.3 This evolution of knowledge on the disease led to the development of several diagnostic criteria Wingerchuk et al1 which were successively revised in 2006,4 then in 2015 (Table 1).5 At the same time, treatment has become more and more codified with a better prognosis. NMO spectrum disorders (NMOSD) represent 1.2% of CNS inflammatory demyelinating diseases.6 Although it is believed to disproportionately affect blacks in mixed populations,7 data on the incidence of NMOSD in black African are scarce. Since Osuntokun in Nigeria in 1970,8 cases of NMO/NMOSD have been reported in black African countries.9 10 11 12 13 14 In Togo, nearly 55% of the population lives in multidimensional poverty, the guaranteed interprofessional minimum wage (GIMW) is 35,000 CFA francs ($US64), while the rate of health insurance coverage is estimated at nearly 5%. We conducted a hospital-based study with the objective of describing possible peculiarities of NMOSD in our setting.
|
Diagnostic criteria for NMOSD with AQP4-IgG |
|
1. At least 1 core clinical characteristic (optic neuritis, acute myelitis, area postrema syndrome: episode of otherwise unexplained hiccups/nausea/vomiting; acute brainstem syndrome; symptomatic narcolepsy or acute diencephalic clinical syndrome with NMOSD-typical diencephalic MRI lesions; symptomatic cerebral syndrome with NMOSD-typical brain lesions) |
|
2. Positive test for AQP4-IgG using best available detection method |
|
3. Exclusion of alternative diagnoses |
|
Diagnostic criteria for NMOSD without AQP4-IgG or unknown AQP4-IgG status |
|
1. At least 2 core clinical characteristics occurring as a result of one or more clinical attacks and meeting all of the following requirements: |
|
a. At least 1 core clinical characteristic must be optic neuritis, acute myelitis with LETM, or area postrema syndrome |
|
b. Dissemination in space (2 or more different core clinical characteristics) |
|
c. Fulfillment of additional MRI requirements, as applicable |
|
2. Negative tests for AQP4-IgG using best available detection method, or testing unavailable |
|
3. Exclusion of alternative diagnoses |
|
Additional MRI requirements for NMOSD without AQP4-IgG and NMOSD with unknown AQP4-IgG status |
|
1. Acute optic neuritis: requires brain MRI showing (a) normal findings or only nonspecific white matter lesions, OR (b) optic nerve MRI with T2-hyperintense lesion or T1-weighted gadolinium enhancing lesion extending over ½ optic nerve length or involving optic chiasm |
|
2. Acute myelitis: requires associated intramedullary MRI lesion extending over 3 contiguous segments (LETM) OR 3 contiguous segments of focal spinal cord atrophy in patients with history compatible with acute myelitis |
|
3. Area postrema syndrome: requires associated dorsal medulla/area postrema lesions |
|
4. Acute brainstem syndrome: requires associated periependymal brainstem lesions |
Abbreviations: AQP4, aquaporin 4; IgG, immunoglobulin G; LETM, longitudinally extensive transverse myelitis; MRI, magnetic resonance imaging; NMOSD, neuromyelitis optica spectrum disorder.
Patients and Methods
This is a multicenter, retrospective study case series diagnosed between 2015 and 2020 in the neurology departments of the Togo university hospital centers (UHC) (Campus, Sylvanus Olympio, and Kara), which constitute the only neurology departments in Togo. The neurology department at Sylvanus Olympio UHC has four neurologists, 18 beds, and an average of 400 hospitalizations and 1,200 consultations per year since 2015. The UHC Campus has four neurologists, 30 beds, and performs an average of 850 hospitalizations and 3,600 consultations per year. The one at Kara UHC has two neurologists, 6 beds, and average 200 hospitalizations and 600 consultations per year since 2015. Patients diagnosed with NMOSD according to the 2015 International Panel for NMO Diagnosis diagnostic criteria (Table 1) were included. The research of anti-AQP4 antibodies was performed at the Cerba laboratory in Paris (IFI on transfected cells). Patients were followed for at least 6 months, and patient progress was assessed with the expanded disability status scale score (Table 2).15 Consent was obtained for the use of the data in the files.
|
Score description |
|
0 Normal neurological exam, no disability in any functional system (FS) |
|
1.0 No disability, minimal signs in 1 FS |
|
1.5 No disability, minimal signs in more than 1 FS |
|
2.0 Minimal disability in 1 FS |
|
2.5 Mild disability in one FS or minimal disability in two FS |
|
3.0 Moderate disability in 1 FS, or mild disability in 3 or 4 FS. No impairment to walking |
|
3.5 Moderate disability in 1 FS and more than minimal disability in several others. No impairment to walking |
|
4.0 Significant disability but self-sufficient and up and about some 12 hours a day. Able to walk without aid or rest for 500m |
|
4.5 Significant disability but up and about much of the day, able to work a full day, may otherwise have some limitation of full activity or require minimal assistance. Able to walk without aid or rest for 300m |
|
5.0 Disability severe enough to impair full daily activities and ability to work a full day without special provisions. Able to walk without aid or rest for 200m |
|
5.5 Disability severe enough to preclude full daily activities. Able to walk without aid or rest for 100m |
|
6.0 Requires a walking aid—cane, crutch, etc.—to walk ∼100m with or without resting |
|
6.5 Requires two walking aids—pair of canes, crutches, etc.—to walk ∼20m without resting |
|
7.0 Unable to walk beyond ∼5m even with aid. Essentially restricted to wheelchair; though wheels self in standard wheelchair and transfers alone. Up and about in wheelchair some 12 hours a day |
|
7.5 Unable to take more than a few steps. Restricted to wheelchair and may need aid in transferring. Can wheel self but cannot carry on in standard wheelchair for a full day and may require a motorized wheelchair |
|
8.0 Essentially restricted to bed or chair or pushed in wheelchair. May be out of bed itself much of the day. Retains many self-care functions. Generally has effective use of arms |
|
8.5 Essentially restricted to bed much of day. Has some effective use of arms retains some self-care functions |
|
9.0 Confined to bed. Can still communicate and eat |
|
9.5 Confined to bed and totally dependent. Unable to communicate effectively or eat/swallow |
|
10.0 Death due to neuromyelitis optic spectrum disorders |
Observations
There were six patients, all Togolese, born and raised in Togo. The mean age was 25.33 years and the sex ratio female/male was 5/1. The mean time from first attack to diagnosis was 122.83 days. At the time of diagnosis, there was isolated spinal cord involvement (2/6) (Fig. 1), simultaneous opticomedullary involvement (3/6), and area postrema syndrome (1/6) (Fig. 2). Five patients were anti-AQP4 seropositive. All six patients had extensive longitudinal myelitis. At 6 months of follow-up, there were one case of death and one case of blindness. Table 3 presents the main data of the six observations.
|
Age/sex/ethnicity |
Clinical feature |
Attacks |
Anti-AQP4 |
Location of lesions |
CSF |
Time from first attack to diagnosis |
Associated pathology |
Treatment of attack |
Background treatment |
Evolution |
|
|---|---|---|---|---|---|---|---|---|---|---|---|
|
Case 1 |
26 yr/F/Ewé |
Cervical myelitis and optic neuritis |
2 in 12 mo |
Positive |
C2 to C7 |
Normal |
12 mo (365 d) |
Viral infection |
Methyl-P Methyl-P Methyl-P Methyl-P Methyl-P Methyl-P |
None |
Death: EDSS = 10 |
|
Case 2 |
28 yr/F/Ewé |
APS then myelitis |
2 in 1 mo |
Positive |
Bulb then C2 |
Normal |
1 mo (30 d) |
Neuromeningeal tuberculosis |
Azathioprine |
EDSS = 0 at 3 yr |
|
|
Case 3a |
30 yr/M/Ewé |
Cervical myelitis and optic neuritis |
2 in 12 mo |
Positive |
C1 to C7 and white matter of the brain |
Normal |
21 d |
None |
Cyclophosphamide |
EDSS = 3 at 4 yr |
|
|
Case 4 |
13 yr/F/Guin |
Cervical myelitis and optic neuritis |
1 since 24 mo |
Negative |
C2 to D2 |
Normal |
1 mo and 12 d |
None |
Azathioprine Azathioprine Azathioprine |
EDSS = 3 at 18 mo |
|
|
Case 5 |
12 yr/F/Ewé |
Cervical myelitis |
3 in 9 mo |
Positive |
C1 to C7 |
Hyperproteinorachia |
8 mo (244 d) |
Viral infection |
EDSS = 7,5 at 6 mo |
||
|
Case 6 |
43 yr/F/Kotokoli |
Cervical myelitis |
1 since 5 mo |
Positive |
C2 to C6 |
Normal |
1 mo and 05 d |
None |
EDSS = 0 at 10 mo |
Abbreviations: anti-AQP4, anti-aquaporin 4 antibodies; APS, area postrema syndrome; CSF, cerebrospinal fluid; EDSS, expanded disability status scale; F, female; M, male; methyl-P, methylprednisolone.
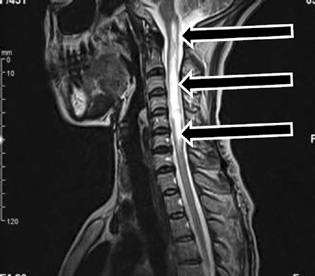
-
Fig. 1 Sagittal fluid-attenuated inversion recovery magnetic resonance imaging sequence demonstrates longitudinally extensive transverse myelitis from C1 to C6 (arrows).
Fig. 1 Sagittal fluid-attenuated inversion recovery magnetic resonance imaging sequence demonstrates longitudinally extensive transverse myelitis from C1 to C6 (arrows).
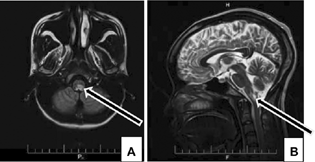
-
Fig. 2 Axial fluid-attenuated inversion recovery (A) and sagittal T2-weighted (B) magnetic resonance imaging sequences demonstrate a bulbar left more lateralized lesion (arrows) in patient with area postrema syndrome.
Fig. 2 Axial fluid-attenuated inversion recovery (A) and sagittal T2-weighted (B) magnetic resonance imaging sequences demonstrate a bulbar left more lateralized lesion (arrows) in patient with area postrema syndrome.
Discussion
The objective of this study was to describe the epidemiological, diagnostic, therapeutic, and evolutionary aspects of NMOSD identified in Togo between 2015 and 2020. As this series only includes patients seen in neurology, there is a risk that it is not exhaustive. Nevertheless, this study reports on Togo's experience with NMOSD.
In 5 years, we have identified six cases of ASNMO in the neurology departments of Togo. This is comparable to the low frequency reported in black African: four cases in 5 years in Mali,10 seven cases in 6 years in Ivory Coast,14 and sixteen cases in 7 years in Senegal.13 This contrasts with some studies that have shown a high prevalence in non-Caucasian populations.7 16 This relative rarity of NMOSD in black African could be related to an underestimation in connection with the difficulties of access to care and the lack of knowledge of health professionals in black African. However, the existence of “protective” factors in this region is not excluded.
The average age of patients was 25.33 years with extremes of 13 and 43 years and two pediatric cases and the sex ratio female/male was 5/1. These data are comparable to the few series from black African.
The mean time from first attack to diagnosis was approximately 4 months (122.83 days) with extremes of 21 and 365 days. The late diagnosis is explained by the lack of knowledge of the disease by the population and the health care personnel. Also, the lack of universal health insurance makes it difficult to perform magnetic resonance imaging (MRI) and anti-AQP4 antibody tests. Indeed, the cost of an MRI scan is at least 1,20,000F CFA or 220 $US (about four times the GIMW). The dosage of anti-AQP4 is performed in Paris, France, at 66,000 F CFA or 120 $US (∼2 times the GIMW).
In this series, relapses were associated with neuromeningeal tuberculosis in case 2, an unlabeled bacterial infection in case 5, and a probable viral infection in case 1. The association with these infectious conditions still makes diagnosis difficult in the context of limited technical facilities.
The acute myelitis that represents the most frequent spinal cord injury in NMOSD was found in five out of six patients at the time of diagnosis. It is found two times more isolated and three times associated with optic neuritis. Isolated optic involvement was not identified, probably due to limited recruitment to patients seen in neurology departments. Thus, the medullary presentation is almost constant and the opticomedullary presentation is frequent, while encephalic presentations such as area postrema syndrome are rare, as reported in Senegal.13
The discovery of AQP4 and its antibody had contributed in understanding the pathophysiology of NMOSD. However, 10 to 25% of patients are anti-AQP4 negative.3 When faced with this situation, the 2015 diagnostic criteria allow the diagnosis to be made if the assay is negative or unknown. The diagnostic criteria make sense in our context where access to anti-AQP 4 assays remains difficult. In this series, five out of six patients are seropositive for the anti-AQP4 antibody. The seronegative case was a 13-year-old child in whom clinical and MRI criteria were met.
Management of NMO/NMOSD attacks is done with corticosteroids, plasma exchange, or immunoglobulins. Our patients received intravenous methylprednisolone in one or more courses depending on the evolution. Almost all of our patients (5/6) were seropositive to the anti-AQP4 antibody and therefore at high risk of recurrence17 18 with the need for background treatment. This treatment can be done either with azathioprine that is more available and manageable in our context. In this series, the evolution was mostly favorable and the sequelae were bilateral blindness, ataxia, paresthesias, and asthenia.
Conclusion
Although rare, NMOSDs are present in Togo. The rarity of NMOSD cases in Togo may be related to underestimation. The clinical presentation is most often classic with a predominance in young girls and frequent medullary involvement. To better describe the NMOSD in the black African population, multicenter and multidisciplinary studies are needed.
Conflict of Interest
None declared.
Funding None.
References
- The clinical course of neuromyelitis optica (Devic's syndrome) Neurology. 1999;53(5):1107-1114.
- [Google Scholar]
- Revised diagnostic criteria for neuromyelitis optica. Neurology. 2006;66(10):1485-1489.
- [Google Scholar]
- International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. 2015;85(2):177-189.
- [Google Scholar]
- Prevalence of neuromyelitis optica spectrum disorder and phenotype distribution. J Neurol. 2009;256(11):1891-1898.
- [Google Scholar]
- Epidemiology of aquaporin-4 autoimmunity and neuromyelitis optica spectrum. Ann Neurol. 2016;79(5):775-783.
- [Google Scholar]
- The pattern of neurological illness in tropical Africa. Experience at Ibadan, Nigeria. J Neurol Sci. 1971;12(4):417-442.
- [Google Scholar]
- New onset neuromyelitis optica in a young Nigerian woman with possible antiphospholipid syndrome: a case report. J Med Case Reports. 2008;2:348.
- [Google Scholar]
- La neuromyélite optique (NMO) au Mali: quelle réalité? Rev Neurol (Paris). 2016;172:A79.
- [Google Scholar]
- First documented case of Devic's neuromyelitis optica in Niger. J African Clin Case Rep Rev. 2017;1(1):18-20.
- [Google Scholar]
- Neuromyelitis optica in Sub-Saharan Africa: the first case report from Togo. Med Sante Trop. 2018;28(2):221-223.
- [Google Scholar]
- Neuromyelitis optica spectrum disorders (NMO-SD) in a Sub-Saharan Africa country: a preliminary study of sixteen Senegalese cases. Mult Scler Relat Disord. 2019;27:179-183.
- [Google Scholar]
- Tanoh Abel Christian, Kohobo C, Dengui S, Kouame D, Aka-Anghui Diarra E, Assi B. Current status of Neuromyelitis Optica (NMO) in Cote d'Ivoire. Afr J Neurol Sci. 2021;40(2):49-58.
- [Google Scholar]
- Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS) Neurology. 1983;33(11):1444-1452.
- [Google Scholar]
- Epidemiology of neuromyelitis optica in the United States: a multicenter analysis. Arch Neurol. 2012;69(9):1176-1180.
- [Google Scholar]
- Neuromyelitis optica positive antibodies confer a worse course in relapsing-neuromyelitis optica in Cuba and French West Indies. Mult Scler. 2009;15(7):828-833.
- [Google Scholar]
- Frequency and prognostic impact of antibodies to aquaporin-4 in patients with optic neuritis. J Neurol Sci. 2010;298:158-162. (1-2):
- [Google Scholar]




