
Translate this page into:
Giant solitary neurofibroma presenting as a neck mass in an infant
Address for correspondence: Dr. Sankalp Dwivedi, Department of Surgery, DMIMS, Sawangi (Meghe), Wardha- 442 004, Maharashtra, India. E-mail: sankalpdwivedi@yahoo.com
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Medknow Publications and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Solitary neurofibroma is a rare tumor of the head and neck region. It is more common in viscera, where it is associated with features of NF1. It occurs most often between the third and fourth decade. These lesions are extremely rare in infants. We report a case of giant, solitary neurofibroma presenting as a progressive mass in parotid region in an infant. This case is unique in its age of presentation (11 months), site, size (about 8 × 15 cm) without any symptoms or neurological deficit.
Keywords
Giant neurofibroma
infant
neck mass
solitary

Introduction
Neurofibromas (NFs) are well-differentiated nerve sheath tumors of benign origin. Mostly they present as a part of systemic disease NF-1 or NF2. Solitary NFs are less common in the head and neck region. These tumors are rarely reported in infants.[1] These lesions present as slowly progressive masses that can either compress to the adjacent vital structures or interfere with normal physiology.[2] We report a case of giant, solitary NF presenting as a progressive mass in parotid region in an infant and review the relevant literature.
Case Report
An 11-month-old female child was admitted in surgery with a progressively enlarging swelling in the right parotid region of two months duration. Her milestones were compatible to her age. Family history was normal. On local examination there was a large (7 × 8 cm), firm, non-tender mass present in right parotid and infra-temporal region with a normal overlying skin [Figure 1]. There were no external features suggestive of NF-1 / NF-2 like macules, lich nodules. There were no suggestive features of compression of underlying neuro-vascular structures. Her hemoglobin was 12 gm % and total leucocyte count and erythrocyte sedimentation rate (ESR) were 6200 / cm m and 20 respectively. The routine urine examination, C-reactive protein, and kidney function test were also within normal limits. Routine X-ray and ultrasonography (USG) of her chest and abdomen and computed tomography (CT) scan of head were normal. The Doppler study of the neck showed a homogenous and hyper-vascular solid mass, situated within the subcutaneous plane of right parotid region. CT with contrast suggested a large well defined homogenous, enhancing mass reaching up to the lateral pharyngeal wall without infiltrating the surrounding structures [Figure 2]. There was no evidence of calcification, cystic degeneration or cervical lymphadenopathy. Fine needle aspiration cytology (FNAC) of the neck was inconclusive. In view of persisting confusion over the diagnosis, surgical exploration was planned. The tumor was exposed and excised completely with extended Blair's incision [Figure 3]. On exploration the tumor was situated within the subcutaneous plane and the parotid gland was shifted superiorly and well separated from the mass. Facial nerve and its branches could easily be separated from the tumor. Postoperative period was uneventful without any residual neurological deficit. Histopathology suggested benign NF [Figure 4]. The patient is doing well at two-year follow-up.
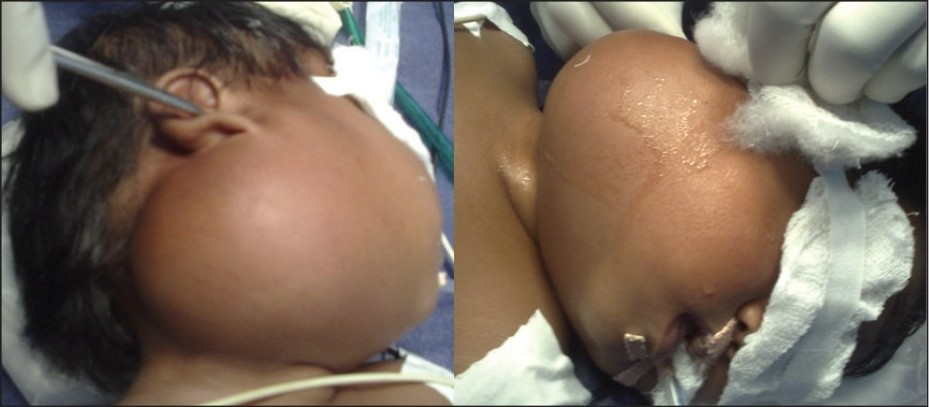
- Clinical photograph showing large mass in the right parotid region
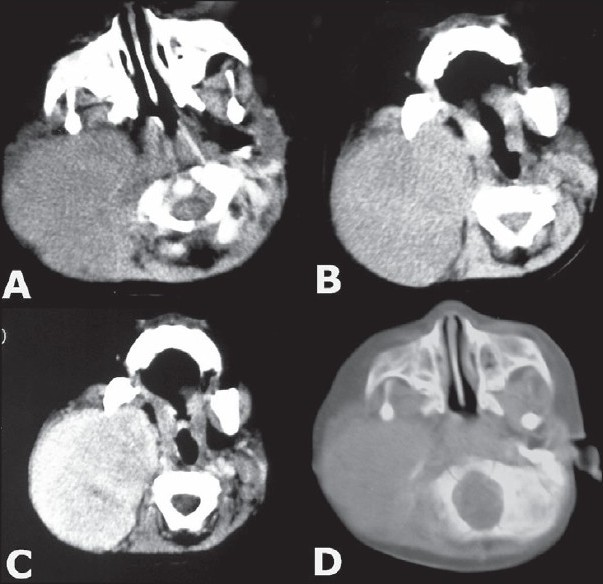
- CT scan showing details of the extent of mass and the lesion was enhancing after contrast administration
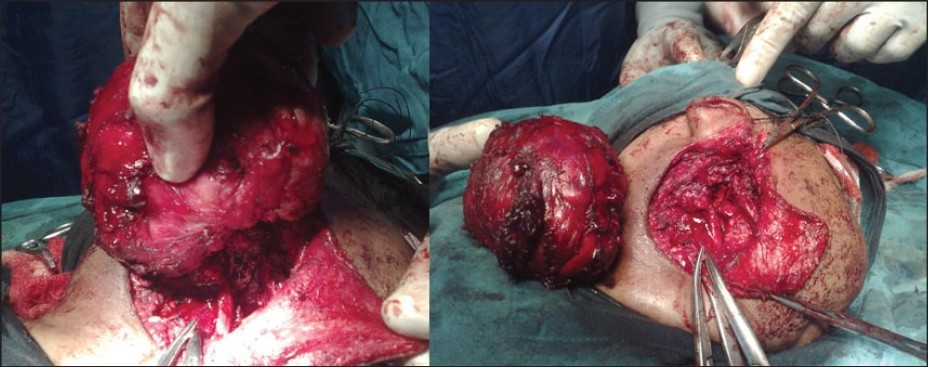
- Intra-operative photograph well defi ned plane of cleavage and complete excision of the tumor
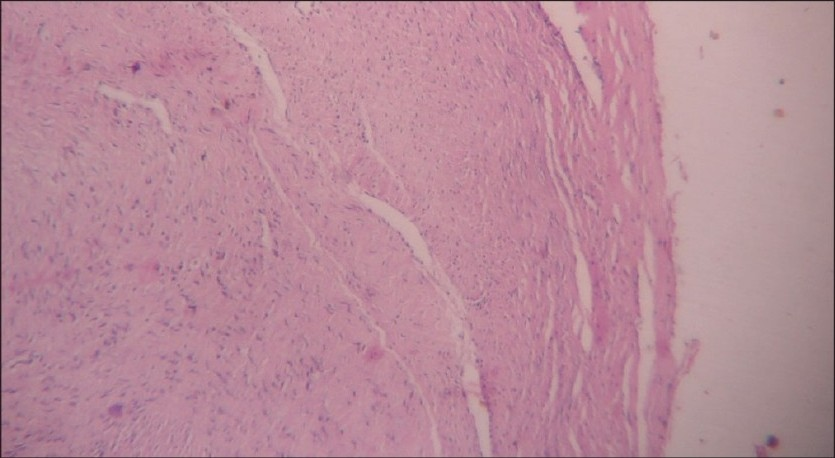
- Histopathology showing features of neurofi broma (H & E, ×40)
Discussion
Peripheral nerve sheath tumors arise from the neural crest. They are mainly NFs, schwannomas and neurogenic tumors. Schwannomas or NFs are most common with a common origin – the schwann cell.[3] In the head and neck, about 25% of all NFs are found but solitary NFs are rare in this region.[1] Generally NFs show autosomal dominant pattern, occurring either due to spontaneous mutation or from the passing of infected gene with complete penetrance and variable expression.[1] They are well-differentiated, non-encapsulated nerve sheath tumor, composed of Shawn cells, perineural-like cells, fibroblasts, and transitional cells and arise within the endo-neurium.[4] The nerve fibers are incorporated within the tumor therefore cannot be separated and resection may sacrifice the nerve of origin.[5] The systemic and hereditary factors are absent in the solitary type. The malignant transformation is extremely low.[6] NFs have to be differentiated from schwannomas and malignant peripheral nerve sheath tumors (MPNST). Schwannomas are encapsulated tumors of nerve sheath and grow eccentrically without infiltrating the nerve and may be dissected out successfully from the nerve. They have a typical palisade pattern of the nuclei, and tumor cell density is higher compared with that in NFs.[6] Mitosis and a mucoid extra cellular matrix with only scattered tumor cells (the indicator of malignancy) are common in schwannoma. MPNSTs are characterized by hypercellularity and pleomorphic tumor cells and nuclei.[6] Histopathologicaly, Type-A tissue of Antoni (the narrow cylinders with tapering ends) - compact and composed bundles of long bipolar spindle cells with oval or rod-shaped nuclei and Type B tissue - a loose texture; and polymorphism of the tumor cells are highly specific structures of neurilemomas.[7] NFs are seen to be slowly growing asymptomatic soft tissue masses and often do not have any neurological deficits. They can be a few millimeters to more than 20 cm in diameter.[7] Plexiform neuromas may be hyper echoic and associated with diffuse thickening of the nerves.[8] The diagnostic workup includes ruling out the presence of other NFs and any possibility of neurofibromatosis NF1 and NF2. The diagnostic criteria for NF1 includes family history, six or more café-au-lait macules, neurofibromas, Lisch nodules, tumor on the optic nerve and skeletal abnormalities, of which two must be present in a patient to be diagnosed as NF1. The diagnostic criteria for NF2 is bilateral vestibular schwannomas (VS) or family history of NF2 (first degree family relative) plus unilateral VS < 30 years or any two of the meningioma, glioma, schwannoma and juvenile posterior sub capsular lenticular opacities/ juvenile cortical cataract.[5] The destruction of bone is common with NF when located within the paranasal sinuses.[6] USG is not very specific and both schwannomas and NFs are commonly homogeneous and hypo echoic and produce posterior acoustic enhancement.[8] CT with contrast in NFs usually shows an ovoid, well-demarcated, homogeneous and iso-attenuation relative to muscle.[36] Heterogeneous contrast enhancement, as demonstrated in the present case, has been described to be typical for NFs.[3] Immuno-histochemically, many of the spindle cells are positive for common neural markers, with patchy staining for CD34 and epithelial membrane antigen. Immuno-reactivity for S-100 protein, neuron-specific enolase, and vimentin is characteristic for NFs.[3] Complete surgical excision is the therapy of choice because it is a benign tumor. Radiotherapy and chemotherapy are considered if there are chances of malignancy or recurrence.[56] Follow-up is mandatory because the lesion may recur. Solitary NFs very rarely undergo malignant change. The transformation of a NF into a MPNST has been observed in 2 - 5% of cases with NF-1.[6]
To summarize, solitary NFs are rare in infants, especially in head and neck region. They may mimic parotid tumor and even with careful evaluation preoperative diagnosis may be difficult. The present case is unique in its age of presentation (11 months), site and size (about 8 × 15 cm), without any symptoms or neurological deficit and could be managed successfully with good outcome.
Source of Support: Nil
Conflict of Interest: None declared.
References
- Sporadic and multiple neurofibromas in the head and neck region: a retrospective study of 33 years. Clin Oral Investig. 2007;11:165-9.
- [Google Scholar]
- Solitary neurofibroma of the inferior nasal turbinate. Auris Nasus Larynx. 1998;25:329-31.
- [Google Scholar]
- Solitary neurofibroma of the palatine tonsil: A case report. Ear Nose Throat J. 2007;86:756-8.
- [Google Scholar]
- Recurrent neurofibroma in the head and neck. J Coll Physicians Surg Pak. 2007;17:629-31.
- [Google Scholar]
- Solitary neurofibroma of the maxillary sinus and pterygopalatine fossa. Otolaryngol Head Neck Surg. 2005;133:458-9.
- [Google Scholar]




