
Translate this page into:
Clinical features and course of glutaric aciduria-Report of six cases
Address for correspondence: Dr. Sadanandavalli Retnaswami Chandra, Faculty Block, Neurocentre, National Institute of Mental Health and Neurosciences, Bangalore - 560 029, Karnataka, India. E-mail: drchandrasasi@yahoo.com
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Sir,
Glutaric aciduria forms a rare group of metabolic diseases with treatment options if diagnosed early. Type 1 has a prevalence of about 1 per 100,000 and characterized by accumulation of 3-OH glutaric acid and presents with hypoglycemia, vomiting, sweaty feet, chorea and failure to thrive.[1] In Type 2 there is accumulation of 2-hydroxy glutaric acid and has a wide spectrum of presentation in clinico-demographic factors and associated with acidosis, hypoglycemia, coma, heart, liver, kidney, pancreas and skull involvement and a late form having a relatively benign course.[234] Type 3 glutaric aciduria is a single peroxisomal enzyme defect causing very long-chain fatty acid deficiency and has got an entirely different clinicoradiological presentation which includes adrenoleucodystrophy and adrenomyeloneuropathies.
Patients with Type 1 and Type 2 are treated with “Well way diet” consisting of low lysine and tryptophan.
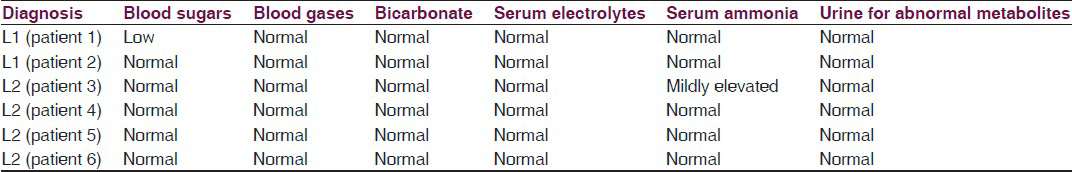
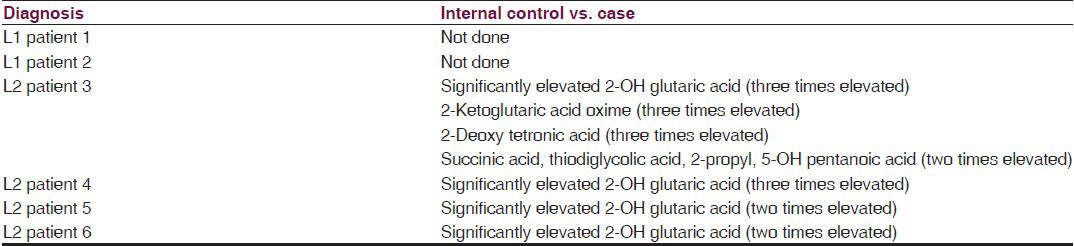
Bread, wheat and wheat products with a daily allowance of 100-150 kcal/kg/day containing low protein with a calorific value of 100-115 kcal/kg/day; protein permitted is 1-1.25 g/kg/day, ready made preparations are available which can be used up to 350 mg/kg/day (max dose 8 g/day) containing l-carnitine, creatine, and glutamine: 100 mg/kg/day each along with riboflavin and alpha-lipoic acid: 10 mg/kg/day each, coenzyme Q10: 8.4 mg/kg/day, pantothenic acid: 5.6 mg/kg/day, alpha-linolenic acid: 150 mg/kg/day, complete pediatric vitamin: 1/2 tablet daily for infants, 1 tablet for young children. Home well-day consists of phenobarbital: 4-6 mg/kg-day titrated to therapeutic drug level and sick-day medications consist of extra dose with anti-inflammatory, antibiotic and anti-emetics. Type 2 glutaric aciduria is treated with prevention of infection, effective maintenance of glycemic levels, supplementation of riboflavin up to 400 mg/day, vitamin C and carnitine.[2] Effective measures are taken to control inflammation, infection, dehydration, electrolyte balance and glycemic control along with managing other system-related co-morbidities when there is encephalopathy. We describe two cases of L1 and four cases of L2 followed up for 3-4 years with characteristic clinical features [Tables 1 and 2], radiological features [Table 3 and Figures 1–4], tandem mass spectroscopy, urine for organic aciduria and chromatography [Tables 4–7 and Figure 5]. The study reveals that L1 glutaric aciduria presents with early, more serious neurodevelopmental and systemic complications and invariably mistaken as infective encephalopathy. L2 can show normal development and late development of seizures and cognitive impairment.[56] Use of specific diet combined with disease-modifying and symptom-modifying treatment reduces morbidity greatly. They have unique radiological features, variable clinical features and are partly treatable. Therefore, a high degree of suspicion is important for diagnosis so that specific treatment can be initiated.


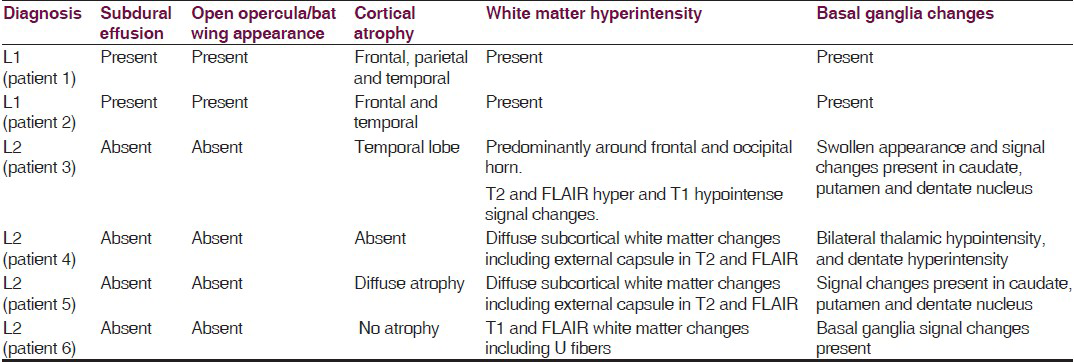
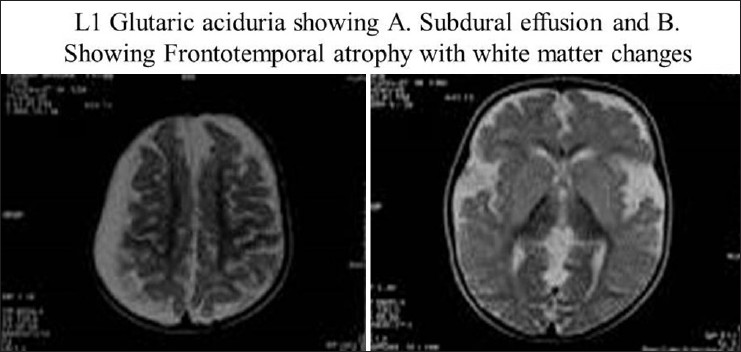
- Open opercula, basal ganglia and white matter changes, sub-dural effusion and atrophy
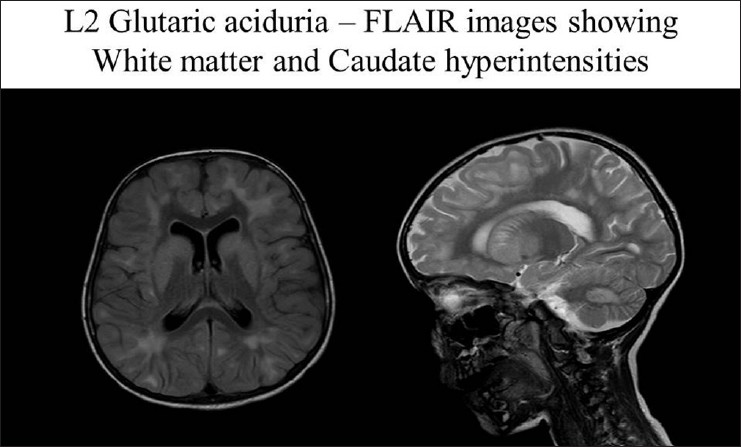
- Periventricular white matter changes and caudate, putamen and dentate nucleus hyperintensities
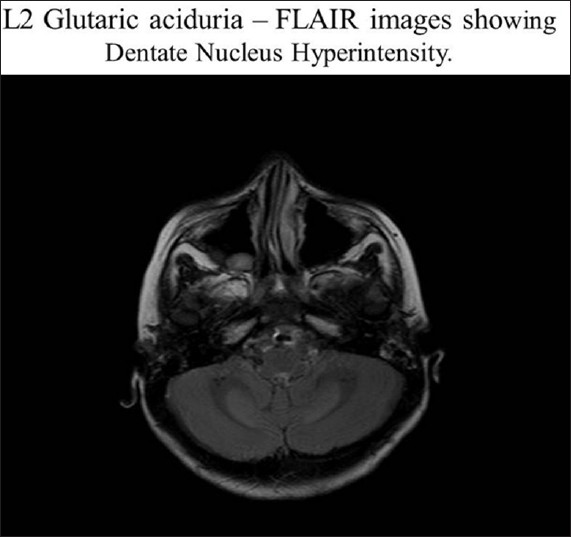
- Dentate nucleus hyperintensities
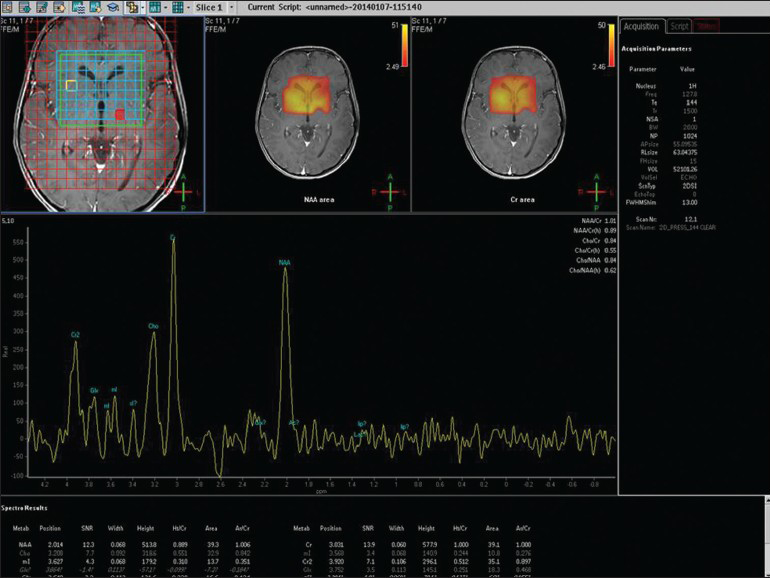
- MRS showing small choline and no lactate peak in L2 glutaric aciduria

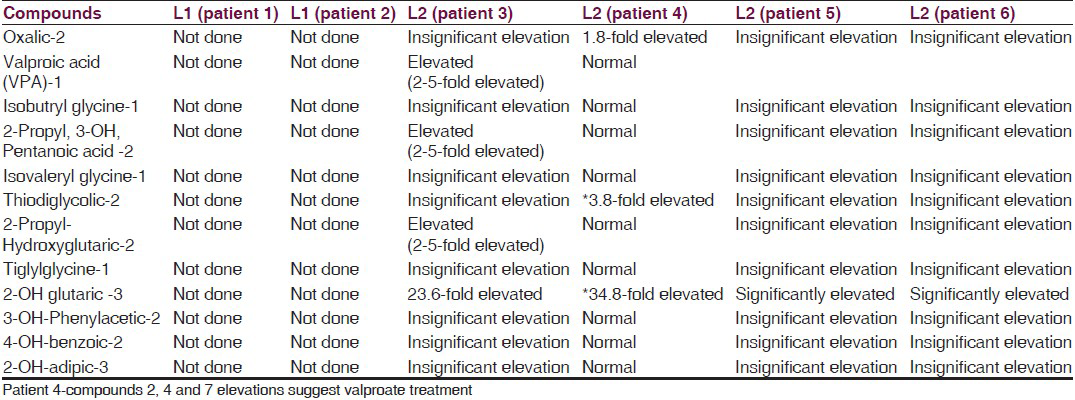
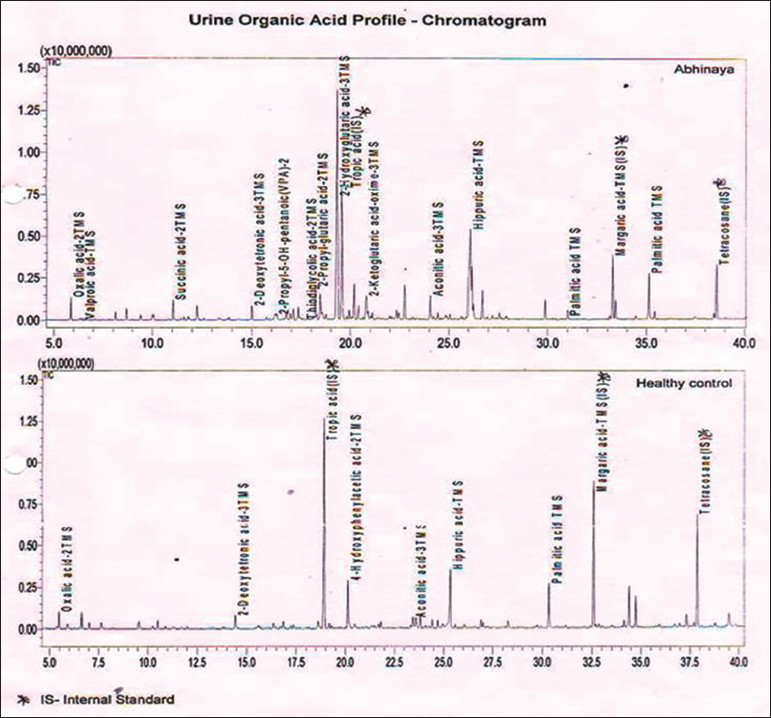
- Urine chromatography showing 2-hydroxy glutaric acid peak
References
- Glutaric aciduria type I: A treatable neurometabolic disorder. Ann Indian Acad Neurol. 2012;15:31-4.
- [Google Scholar]
- Type I glutaric aciduria, part 1: Natural history of 77 patients. Am J Med Genet C Semin Med Genet. 2003;121c:38-52.
- [Google Scholar]
- Genetics home reference. Available from: http://www.ghr.nlm.nih.gov/condition/glutaric-acidemia-type-ii/show/References
- [Google Scholar]
- Risk of sudden death and acute life-threatening events in patients with glutaric acidemia type II. Mol Genet Metab. 2008;93:36-9.
- [Google Scholar]
- Progress in understanding 2-hydroxyglutaric acidurias. J Inherit Metab Dis. 2012;35:571-87.
- [Google Scholar]
- Cerebral neoplasms in L-2 hydroxyglutaric aciduria: 3 new cases and meta-analysis of literature data. AJNR Am J Neuroradiol. 2012;33:940-3.
- [Google Scholar]




